中成药上市后临床安全性评价核心数据集的构建方法探索*
2019-01-29邱瑞瑾胡嘉元何天麦韩松洁张晓雨关曼柯商洪才
邱瑞瑾,李 敏,胡嘉元,黄 涯,何天麦,韩松洁,郑 蕊,张晓雨,关曼柯,陈 静,商洪才**
(1.北京中医药大学东直门医院中医内科学教育部和北京市重点实验室 北京 100700;2.天津中医药大学附属保康医院 天津 300193)
随着中成药的应用日渐增多,其安全性问题越来越凸显。2017年国家药品不良反应监测年度报告显示,中药不良反应占16.1%,其中严重不良反应/事件报告占10.6%[1]。由于严重不良反应/事件,2017年有两种中成药被国家食品药品监督管理总局召回和暂停销售。
上市前研究时间有限,难以观察到中成药的迟发不良反应,加上样本量较小,难以及时发现罕见不良反应,因此大部分中成药说明书安全性信息不足,除不良反应外,注意事项、禁忌、药物间相互作用、药理毒理、特殊人群(如儿童、孕妇及哺乳期妇女、老人等)用药以及用药时间、用法用量等安全性信息标识较少[2-5]。有时即使标识了相关项目,也可能并无实质内容。研究显示,中成药说明书中不良反应标识达58.3%,但其中35.7%的内容为“尚不明确”、“如与其他药物同时使用可能会发生药物相互作用,详情请咨询医师或药师”或“尚无本品与其他药物相互作用的信息”等[2]。这些内容无法有效指导临床安全性用药,因此开展中成药上市后临床安全性再评价,提供中成药临床安全性证据,修改与完善中成药说明书刻不容缓。
自2009年国家食品药品监督管理总局发布《关于做好中药注射剂安全性再评价工作的通知》以来,相继有研究者探讨了中药上市后安全性医院集中监测技术规范[6]、中成药上市后临床安全性评价研究模式的建立[7]、中药注射剂临床安全性集中监测研究设计与实施专家共识等[8],为中成药上市后临床安全性再评价的规范化研究指明了方向。
中成药上市后临床安全性再评价的目的是明确药品危险因素,进而为制定风险管控计划,指导临床合理用药,降低风险提供科学依据[9]。中成药上市后风险来源有多种,包括药品质量风险、不良反应风险、不合理用药风险等[10]。其中不良反应风险又包括与剂量相关的不良反应、与剂量无关的不良反应、联合用药引起的不良反应、非预期的不良反应等。临床安全性再评价的方法和数据来源也较多,如随机对照试验、病例对照研究、病例报告、病例系列、队列研究、医院集中监测、自发呈报等,由于研究方法不同,需要采集和分析的临床安全性数据有一定差异,加上中成药普遍存在漏报现象,在一定程度上对研究结果的真实性产生影响。
为确保中成药上市后临床安全性再评价能得到相对真实、全面的结果,除了需要制度和法规的保障之外,也需要在研究方法上进一步完善,尤其在数据采集方面。为减少不同研究在临床安全性数据的采集和报告方面的差异,使更多研究能在系统评价中进行合并和同类比较,为中成药的安全性应用提供更高级别的证据,同时也为中成药说明书的修订和完善提供依据,笔者结合临床安全性评价的特点,初步提出构建中成药上市后临床安全性评价核心数据集(Core data set)的构建方法。
核心数据集是指特定疾病领域临床实践或临床研究中应当采集的标准化的数据集合[11],是核心结局指标集(Core outcome set)概念的延伸和扩展。与核心结局指标集相比,核心数据集纳入的范围更加广泛,除了结局指标之外,其它能反映疗效或安全性的指标,如年龄、性别、吸烟史、合并用药、患病时间等相关的指标也可以纳入。
构建中成药上市后临床安全性评价核心数据集有以下几方面的意义:①减少不同研究采集或报告的数据类型的差异,使更多研究能纳入系统评价/meta分析,为临床安全性使用中成药提供更高级别的证据;②规定中成药临床安全性评价中应采集的数据,有利于及时发现并减少不良反应漏报的情况,使研究结果更能反映真实情况;③通过对相关暴露因素的数据采集,可进行不良反应关联性分析;④通过规定临床安全性评价中应采集的核心数据,可以减少由于信息繁冗造成的研究者依从性差的情况。
在团队构建核心结局指标集的经验和基础上[12,13],笔者进一步探索中成药上市后临床安全性评价核心数据集的构建方法,为中成药临床安全性再评价数据采集条目的选择提供方法学借鉴。
1 确定中成药临床安全性评价核心数据集的适用范围
中成药临床安全性评价采集的数据与目标疾病、目标人群、研究目的、给药途径、评价方法等密切相关,因此临床安全性评价核心数据集的适用范围应从以下几个方面考虑:
1.1 目标疾病
不同疾病在进行临床安全性评价时,数据采集的侧重会有所不同。因此,构建临床安全性评价核心数据集应首先明确目标疾病。
1.2 目标人群
目标人群不同可能会造成风险因素的差异,加上目前中成药说明书中对特殊人群(儿童、老人、经期/怀孕/哺乳期女性等)的安全性信息研究不足,因此临床安全性评价核心数据集应明确适用的人群范围,如全部人群或特殊人群。
1.3 研究目的
临床安全性评价的目的包括几个方面,如明确不良反应发生率、发现新的不良反应、评价长期用药的安全性或探讨不良反应的相关危险因素等。研究目的不同,需要采集的数据会存在一定的差异,如研究目的是评价不良反应发生情况或长期用药的安全性时,需要收集不良反应事件以及实验室指标如血常规、生化指标等。而探讨不良反应的相关危险因素时,所有可能相关的暴露因素,如年龄、性别、疾病史、吸烟史、饮酒史等均应考虑。因此,在研究开始前应明确临床安全性评价核心数据集所适用的研究目的,避免研究缺乏针对性。
1.4 给药途径
不同给药途径所造成的不良反应发生率和不良反应的危险因素会有很大差异。发生不良反应最常见的两种给药途径是静脉注射和口服,分别占54.0%和39.4%[1]。从危险因素的角度看,中成药注射剂的不良反应可能与溶媒类型、配药时间、滴速、是否冲管、合并用药等相关[14],而其它给药途径不存在这些因素。因此中成药临床安全性评价核心数据集的适用范围应考虑不同给药途径的差异。
1.5 评价方法
评价中成药临床安全性的方法有很多,包括随机对照试验、医院集中监测、队列研究、病例对照研究、病例系列、病例报告等,每种研究又有各自的特点。如随机对照试验采用随机分组的方法,尽可能地避免混杂因素的影响,但由于其严格的纳入排除标准,导致结果可能会和真实情况有一定差异,并且临床安全性往往作为次要结果进行评估;医院集中监测属于真实世界研究范畴,能在短时间内较全面地认识药物安全性,是药品上市后再评价的重要手段和主要内容,不仅能前瞻、客观、定量分析中成药的不良反应或不良事件,还可以发现新的不良反应风险信号,确定危险因 素[8,15-17];队列研究可用于评价干预措施的长期不良反应;病例对照研究能同时探索多种暴露因素和不良反应之间的关联性,尤其在评价罕见不良反应或潜伏期很长的不良反应方面有明显优势;病例系列和病例报告能为发现不良反应提供基础的依据。
不同评价方法有各自的利弊,在采集信息方面也有区别,因此在进行临床安全性评价核心数据集构建时,其适用范围应考虑不同研究方法的区别,如适用于试验性研究或观察性研究。
2 研究方案注册及伦理审查
在确定中成药安全性评价核心数据集的适用范围之后,应制定研究方案。为减少偏倚,保证研究的透明化及参与者的权益,研究方案应通过伦理审查并进行注册[18,19]。一般而言,此类研究可在“有效性试验核心结局测量”(Core Outcome Measures in Effectiveness Trials,COMET)行动的数据库进行注册(http://www.comet-initiative.org/)。
3 形成专家指导委员会
在正式研究前,应由相关领域的专家组成专家指导委员会。专家指导委员会可由5-7人组成,应包含不同的利益相关群体,如临床医生、临床研究者、方法学专家、杂志编辑、政策制定者、企业代表等。专家指导委员会有几个方面的作用:①在数据的收集和完善的过程中给予指导和建议;②促进不同利益相关群体的专家参与研究;③参加共识会议,促进共识的达成;④确定最终的核心数据集。
4 形成中成药临床安全性评价原始项目清单
在完成上述工作后,需要通过两种方式形成中成药临床安全性评价原始项目清单:
4.1 文献研究
文献研究主要是根据临床安全性评价核心数据集的适用范围,对相关研究进行系统评价,提取相关的临床安全性指标及其它数据。文献研究应尽量全面,除包括试验性研究和观察性研究外,也可从药品不良反应监测系统中提取相关信息。通过文献研究可初步形成临床安全性评价原始项目清单。
临床安全性评价原始项目清单应根据核心数据集的适用范围及研究目的提取。如核心数据集用于评价长期用药的临床安全性,则不良反应发生的时间,不良反应的类型、程度及预后,应纳入清单;若核心数据集是用于不良反应关联性评价,则任何可能相关的暴露因素均应纳入,包括联合用药情况、证候、疗程、不良反应发生的时间、药物的用法用量、患者年龄、性别、病情及进展、吸烟史及饮酒史等,以便为进行关联性分析,识别风险提供依据。在完成数据提取后,应根据数据的类型进行分类,如分为人口学特征、药物用法用量、实验室指标、不良反应/事件等。
4.2 半结构化访谈
通过文献研究形成的临床安全性评价原始项目清单未必能全面覆盖其适用范围,也不能全面反映所有利益相关群体的意见,因此,应根据清单的情况,对专家指导委员会的专家进行半结构化访谈,根据专家意见进一步补充清单。
此外,在循证医学的研究和实践中,越来越注重患者的参与,加上临床安全性评价的对象主要是患者,因此患者的观点在临床安全性评价核心数据集的形成中至关重要。可以通过半结构化访谈、焦点小组的方式获得对患者重要的数据,补充到原始项目清单中。
5 德尔菲调查
德尔菲调查是采用背靠背的方法咨询专家意见,通过多轮次的反馈修正,使专家意见趋于一致,得出预测结果的一种评价方法[20]。德尔菲调查已在核心数据集的研究中成功实施[11]。
德尔菲调查一般进行2-3轮,参与者来自不同利益相关群体。在中成药临床安全性评价核心数据集中,应包括临床医生、研究者、方法学专家、杂志编辑、政策制定者、企业代表等不同专家参与,使核心数据集能在不同利益相关群体中达成共识。
德尔菲调查中不同利益相关群体的选择应具有代表性,包括地域、年龄、性别、资历、教育经历等方面。然而,每个利益相关群体样本量的确定目前尚无统一的方法,一般而言,参与者越多越好[21]。
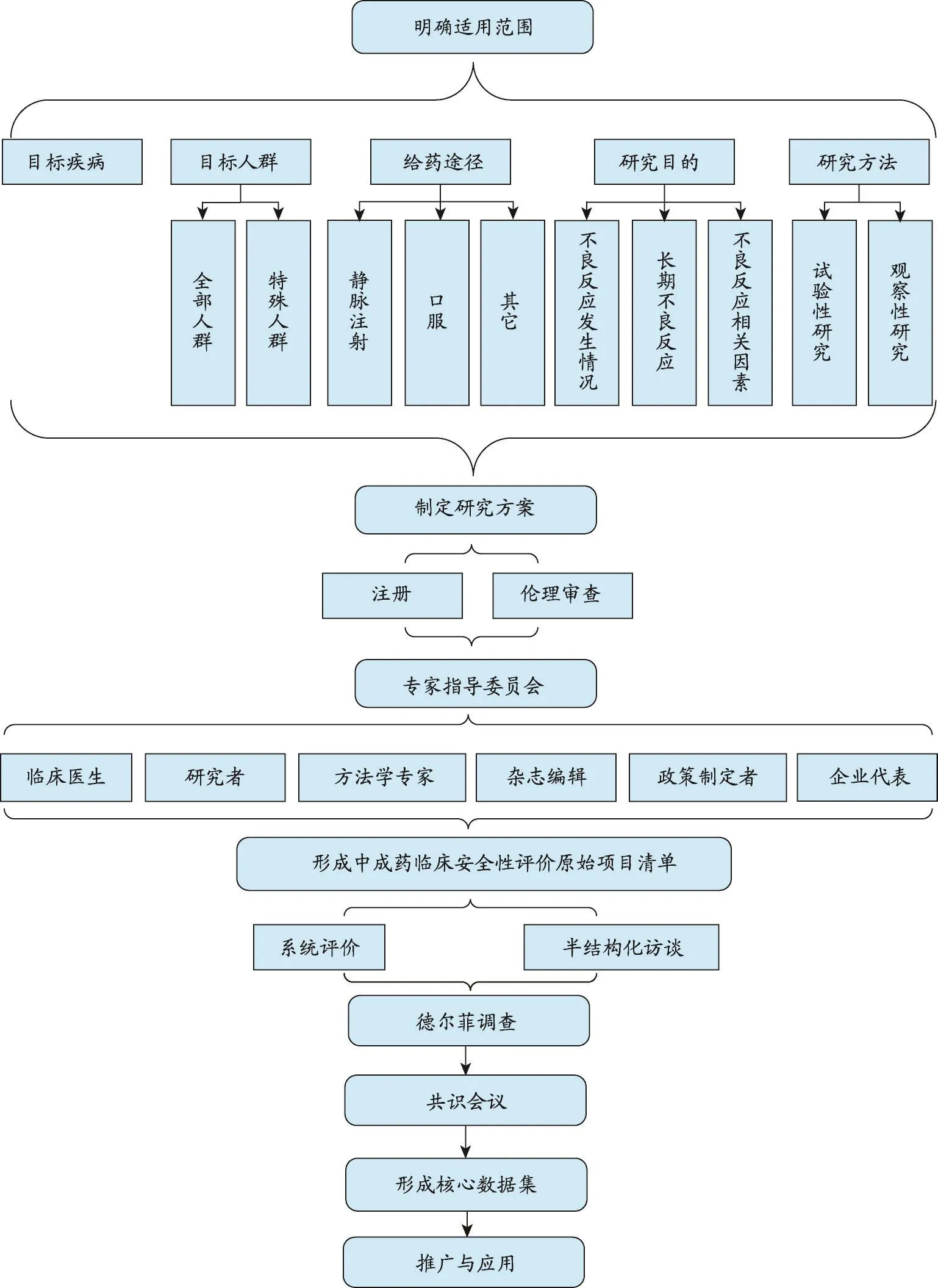
图1 中成药临床安全性评价数据集构建流程
德尔菲调查问卷一般采用是GRADE(Grading of Recommendations Assessment,Development and Qualitative research)工作组推荐的9分Likert评分法,1-3分代表不重要,4-6分代表重要,7-9分代表非常重要[22]。通过专家打分,来判断哪些项目可以纳入核心数据集。
此外,在德尔菲调查中,有很多因素可能影响研究结果[23],因此,研究者应严格遵循研究方案进行,尽量避免在研究过程中更改方案而造成结果偏倚。在制作德尔菲调查问卷时,不同利益相关群体的专家最好能参与调查问卷的设计,确保调查问卷结构清晰,语言通俗易懂,并进行预调查以保证调查问卷的可理解性和有效性。研究过程中,研究者应采取各种途径如电话、邮件、短信提醒或其它激励措施减少偏倚,并在研究结束之后进行失访偏倚评估。
德尔菲调查的目的是使专家意见趋于一致,因此,在完成2-3轮的调查后,应对各项目达成共识的程度进行评估。由于共识的定义、达成共识的程度目前并不统一,因此研究方案中应予以明确规定并避免在研究过程中更改。
6 共识会议
在2-3轮德尔菲调查完成之后,专家指导委员会应对结果进行审核,并邀请不同利益相关群体的专家参加共识会议。共识会议的专家人数不应过少,以免不同利益相关群体的专家无法充分发表意见,但人数也不应过多,以免难以达成共识。共识会议主要对德尔菲调查中未达成共识的项目进行讨论,并再次进行打分,确定最终纳入核心数据集的项目。
7 推广及应用
中成药临床安全性核心数据集完成后,研究者应通过多种途径进行推广,如论文、会议、网络等,使核心数据集能真正应用到中成药临床安全性评价中,减少临床安全性指标漏报的情况,为中成药的临床安全性应用和中成药说明书的修订及完善提供证据。
8 总结
经过二三十年的发展,核心结局指标集在某些领域的应用取得了明显的效果[24]。核心数据集作为核心结局指标集概念的扩展及延伸,虽然目前研究并不多,但由于其纳入的项目并不仅限于结局指标,也可以纳入一般的人口学特征及其它危险因素,因此比较适用于中成药临床安全性评价。在中成药临床安全性评价中使用核心数据集,可能会帮助研究者减少项目漏报的情况,通过比较核心数据集的报告内容能更容易判断不同研究异质性的来源,同时也可以减少同类研究中报告项目的异质性,使更多研究能进行同类比较或合并分析,此外,全面的数据采集也有助于深入研究中成药安全性的风险来源,为中成药的安全性应用提供证据。中成药临床安全性评价数据集的构建流程见图1。
