ANKRD11基因突变所致 KBG综合征1例并文献复习
2019-01-25李秋月章淼滢罗飞宏
李秋月 杨 琳 吴 静 陆 炜 章淼滢 罗飞宏
1 病历资料
女,7岁8月,因“身材矮小”于2017年8月28日至复旦大学附属儿科医院(我院)内分泌遗传代谢科就诊。
现病史:患儿自生后即较同龄儿生长缓慢,来院前1年家长述身高增长约4 cm。我院门诊测身高111.0 cm(<-3SD)、体重17.7 kg(<-2SD),骨龄4岁,拟诊 “矮小症”。患儿15月余独立行走,2岁余说话。平素神情冷漠、寡言少语、拒绝与陌生人交流,偶脾气暴躁,学习成绩差。既往无特殊慢性疾病史。患儿平素饮食欠佳、挑食,睡眠和二便未见异常。
出生史及家族史:患儿系G1P1,足月顺产,出生身长48 cm,出生体重2 400 g。父亲身高174 cm,母亲身高156 cm,否认近亲结婚和家族性矮小病史。
体格检查:T 36.7℃,P 75·min-1,R 22·min-1,BP 90/62 mmHg。神志清楚,精神反应可,呼吸平稳,皮肤无黄染、皮疹、出血点、瘀斑,皮肤弹性可。患儿三角脸、宽眉、鼻梁宽、人中平、上唇呈帐篷样、下颌小、双侧耳大且前倾,上颌中切牙大、有缝隙(图1A);双手小指侧弯(图1B);髋内旋明显大于外旋,“内八字”脚(图1C);脊柱侧凸(图1D,E),心、肺听诊未见异常,神经反射正常,女性外生殖器、双侧乳房B1期,阴毛Turner 1期。
实验室检查:甲状腺功能、皮质醇、糖类抗原CA125、CA199无异常;肝、肾功能、糖耐量实验及糖化血红蛋白含量均未见异常;生长激素(GH)激发试验,生长激素峰值5.7 ng·mL-1(非GH缺乏儿童峰值≥10 ng·mL-1),胰岛素样生长因子(IGF-1)271(参考值57~316)ng·mL-1,胰岛素样生长因子结合蛋白3(IGFBP3) 4.6(参考值1.6~6.5)μg·mL-1。
影像学及辅助检查:X线示骨龄7岁(图1C),脊柱侧凸(图1D,E)。颅脑MRI未见明显异常。韦氏儿童智力量表(WISC-R)测试总分56,其中语言总分55,操作总分72。口、眼、耳检查未见异常。
为进一步明确诊断,经患儿父母知情同意后行家系外周血全外显子基因测序(WES)。依据我院分子诊断中心数据分析流程,结合 WuXi Next CODE 分析软件(CSE)分析测序数据;通过 Burrow.Wheeler Aligner(BWA)与NCBI RefSeq进行匹配比对;采用ANNOVAR和VEP软件以及注释程序注释变异数据。包括用NCBI RefSeq和SwissPort进行基因注释。用HGMD、OMIM 和ClinVar进行疾病相关注释。用于人基因组计划、EVC6500、ExAC和我院分子诊断中心已建立的内部数据库进行突变频率注释;用SIFT、Polyphen 2和MutationTaster进行突变预测。通过突变频率和变异类别的筛选及与疾病的相关关系筛选出候选突变。
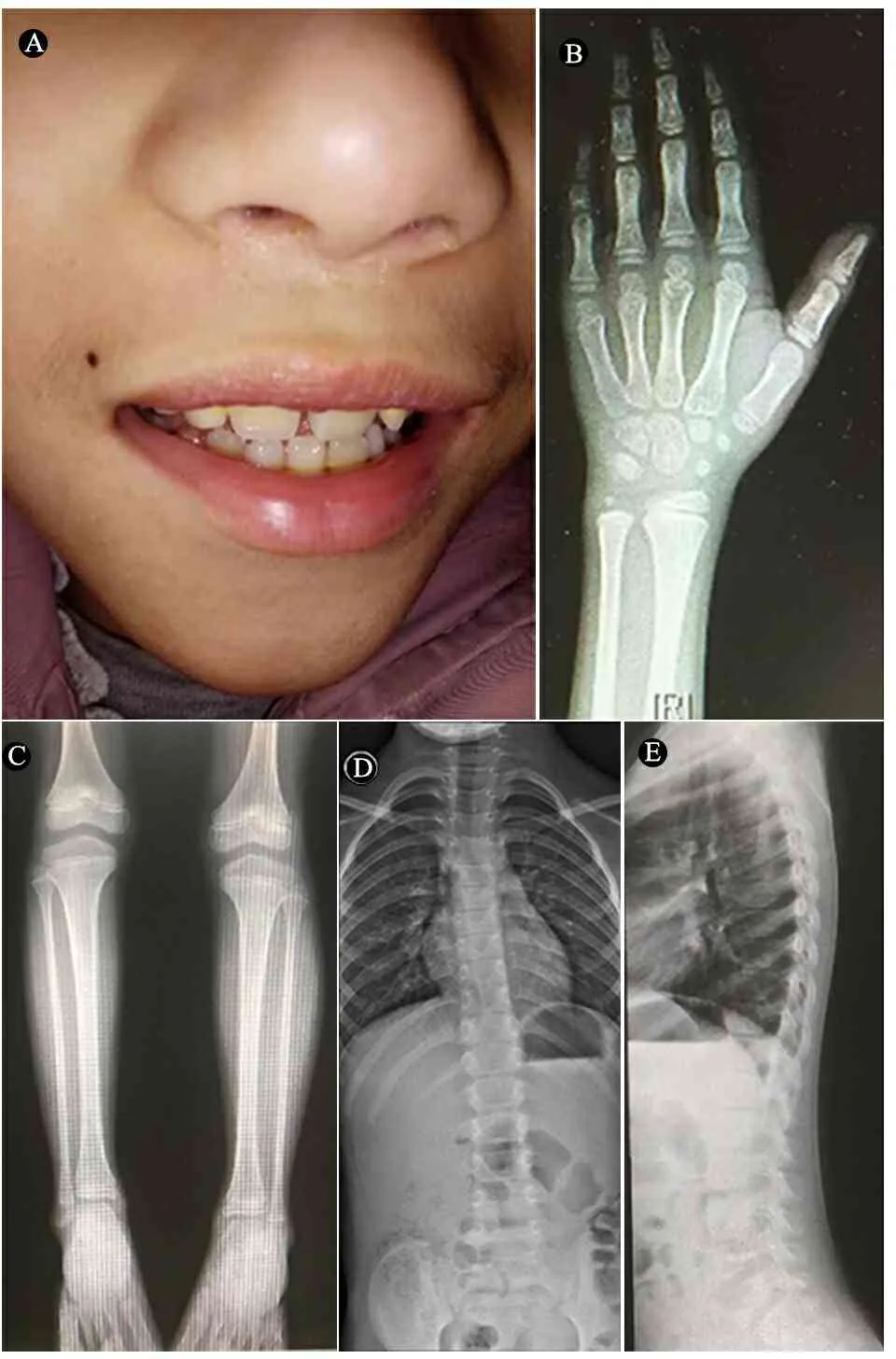
图1患儿颅面部及骨骼表现
注 A:三角脸、人中平、上唇呈帐篷样、下颌小、双侧耳大且前倾,上颌中切牙明显增大;B:手指短小、末指弯曲,骨龄7岁;C : 内八字脚;D,E:脊柱侧凸
WES结果提示(图2):患儿ANKRD11基因(NM_013275)9号外显子存在1个杂合突变c.6972dupC(p.P2271Pfs*8),对患儿及其父母进行Sanger验证,引物F:CCTCCAGAAGAGA TGCCTCC,R:TGGTCATGCGCTGAGGG-ATC,患儿该位点出现套峰,以后序列出现移码突变,其父母均未出现该位点的突变峰。
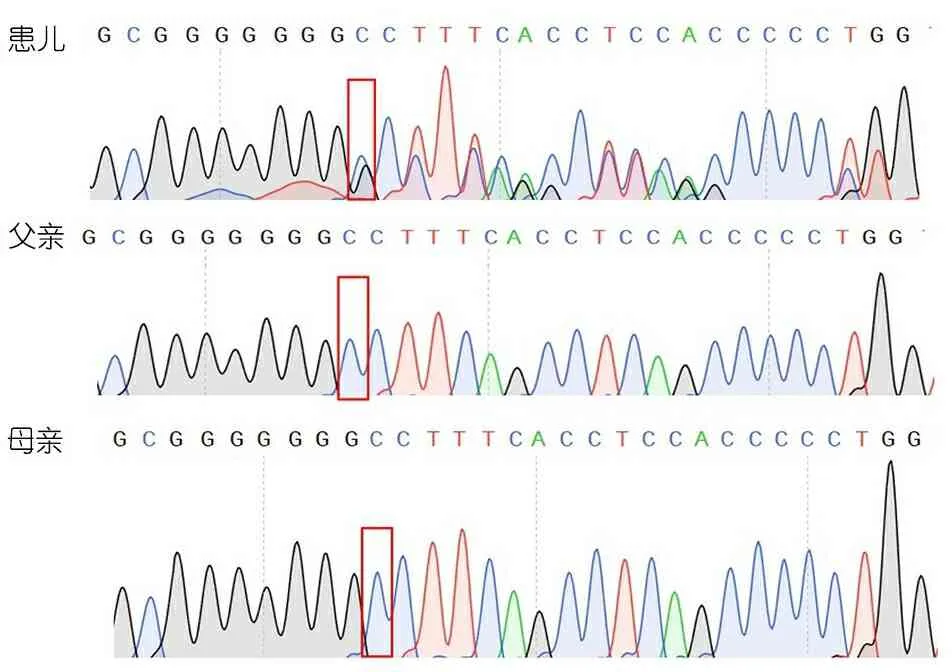
图2患儿及父母测序图谱
Mutation Taster软件对突变蛋白的功能预测引起疾病,根据基因测序结果并结合患儿临床表现,确诊为KBG综合征(macrodontia, mental retardation, characteristic faces, short stature, and skeletal anomalies,KBGS)。
因重度生长落后及GH缺乏,采用外源生长激素治疗,随访至2018年9月,生长速率每年5 cm,具体诊疗经过见图3。
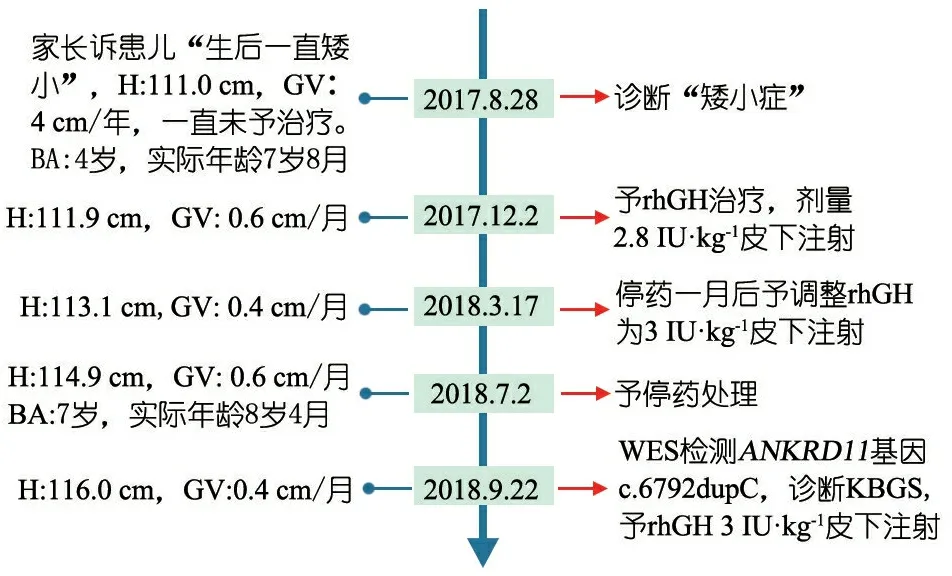
图3临床诊治时间轴
注 H: 身高;GV:生长速率;BA:骨龄;rhGH:重组型生长激素
2 文献复习
以“((((((ankyrin repeats containing cofactor 1) OR ankyrin repeat domain 11 protein) OR ANCO-1 protein) OR ANKRD11) OR 16q24.3 microdeletion ) AND (((((Short stature) OR characteristic facies) OR macrodontia) OR mental retardation) OR skeletal anomalies)OR KBG)” 为检索式在Pubmed、Web of science数据库检索,以“KBG综合征”、“ANKRD11”、“16q24.3微缺失”为关键词在万方数据库和中国知网数据库检索,检索时间均从建库至2018年10月31日。未检索到ANKRD11突变所致KBGS的中文文献,英文文献132篇,逐篇阅读筛选后,22篇英文文献报告了154例KBGS[1~18,21,22,25,26],来自欧洲[1~5,8~10,12~18,22]和亚洲[6,7,11,26],涉及140种突变类型,其中90种突变已包含于HGMD数据库(Human Gene Mutation Database)。加上本文报告1例共155例,发病年龄1~66岁,其中ANKRD11基因突变114例,16q24.3微缺失型41例。表1汇总两种类型KBGS临床表型。
表1显示KBGS以颅面部、骨骼系统、中枢神经系统表现最常见。①颅面部异常主要表现为:三角脸、发际线低、一字/连眉、鼻梁宽、鼻孔前倾、人中长/平、上颌中切牙过大等;②神经系统异常主要表现为:语言发育迟缓、智力不同程度低下及学习障碍;③骨骼系统主要表现为:小指侧弯,不同程度的身材矮小、骨龄延迟,前囟闭合延迟。先天性心脏病也在多例患儿中出现;髋关节畸形、大脑及小脑异常仅在个别病例中报道。男性和女性两组临床表现三角形脸(71.1%vs. 37.8%)、一字/宽眉(69.4%vs. 42.3%)、眼距宽(59.0%vs. 36.4%)、椎体异常(50.0%vs. 20.5%),差异均有统计学意义。

16q24.3微缺失型患儿经典的颅面部异常(70.6%)、手部异常(50.0%)发生率较ANKRD11基因突变型低,但自闭症、先天性心脏病、隐睾的发生率较ANKRD11基因突变型高;前囟闭合延迟(10%)仅在ANKRD11基因突变型中报道;ITP(12.2%)仅在16q24.3微缺失型中报道(图4)。
3 讨论
KBGS(OMIM # 148050 KBG SYNDROME,KBGS)于1975年首次由Herrmann, Pallister, Tiddy和Opitz报道[23],以广泛发育迟缓(语言发育迟缓尤为突出)、颅面异常、上颌中切牙过大和骨骼异常为特征的罕见常染色体显性遗传性疾病,常见的器官异常还包括室间隔缺损、听力丧失、隐睾或生殖器发育不全等[18]。国内尚未有KBGS报道,本文女性患儿经WES发现ANKRD11基因(AD)c.6792dupC杂合突变,在c.6792位插入一个胞嘧啶碱基,使第2271位起后第8位编码天冬氨酸的密码子突变为终止密码子,导致肽链的合成提前终止,为比较明确的致病突变;本文报告病例具有典型的KBGS临床表型,颅面部表现,如三角脸、鼻梁宽、前发际线低等,发育迟缓,矮小,小指侧弯、内八字脚,智力低下及学习障碍,为国内首次报道的病例。
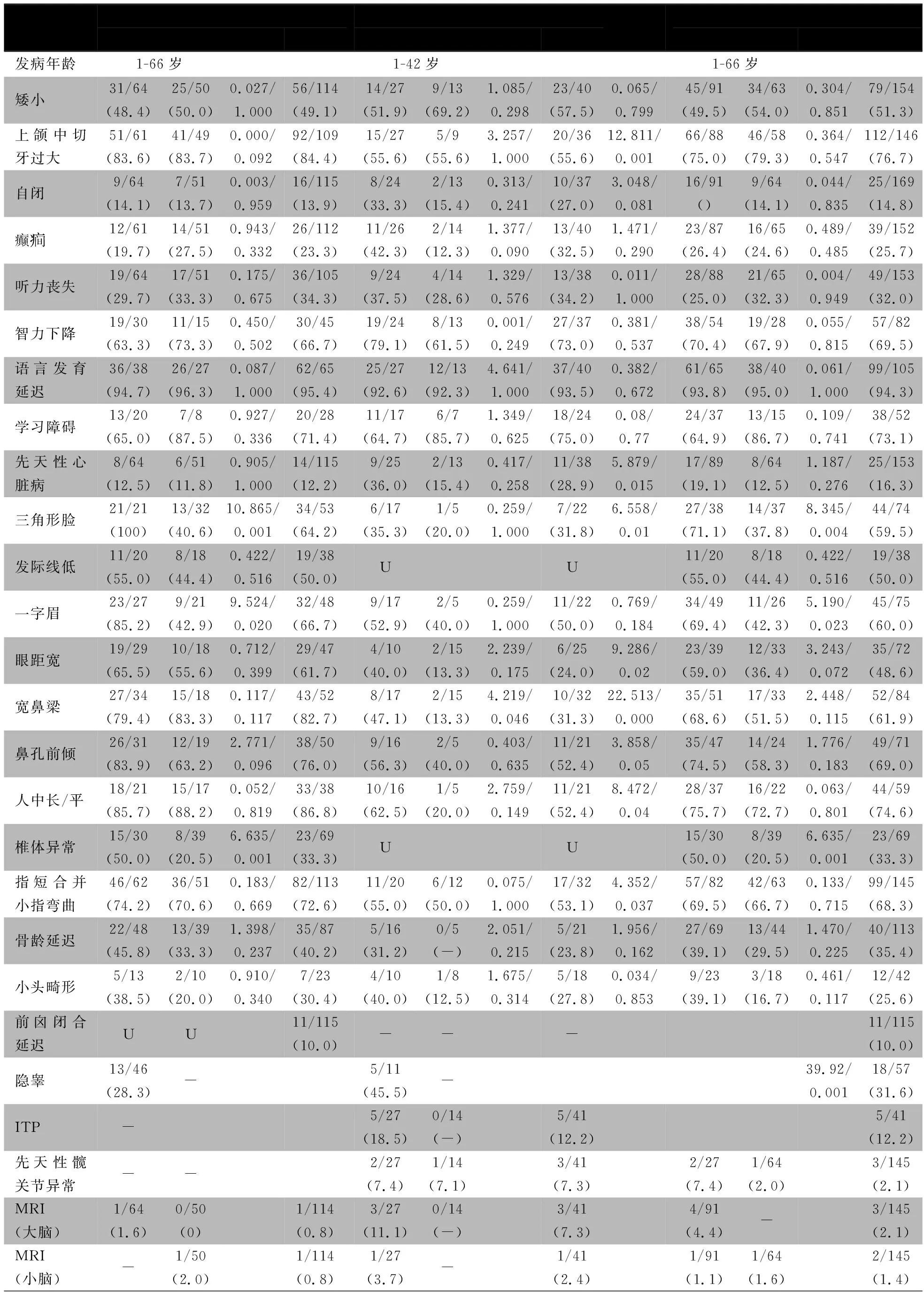
表1 ANKRD11基因突变型与16q24.3微缺失型KBGS临床表现
注 U:未提及;-:未发生;ITP:特发性血小板减少性紫癜
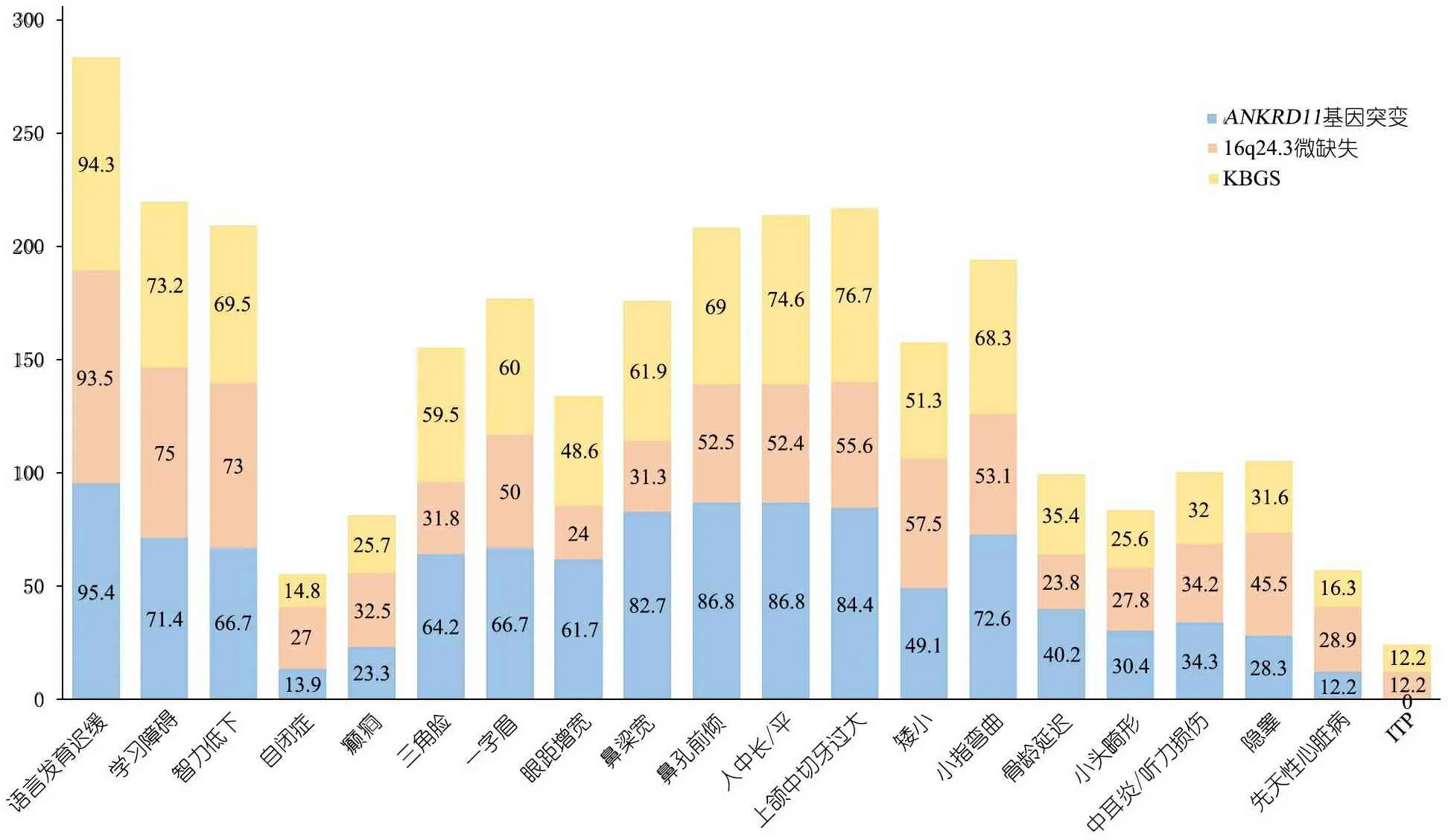
图4ANKRD11突变型、16q24.3微缺失型及KBGS各表型之间对比
部分KBGS患者颅面部表型一般不以某种特殊面部表型单独存在,相对容易诊断,但是另一部分KBGS与其他疾病有重叠的表型,诊断时应仔细鉴别。①Coffin-Siris综合征(CSS):是一种罕见的以生长发育迟缓、智力低下、颜面粗糙、小指指甲缺失及第5指骨(或跖骨)远端缺失的先天性畸形。CSS与KBGS在第五指甲缺失及第5指骨(或跖骨)远端缺失表现明显不同,前者十分常见,但在KBGS中未见相关报道。②Cornelia de Lange综合征(CdLS):主要表现为颅面部异常,如多毛、发际线低、连眉、长睫毛、宽齿缝或缺牙、鼻梁宽、鼻孔前倾、长且突出的人中、口角下垂、小或方形的下颚等,患者常有产前发育迟滞。CdLS与KBGS在上颌中切牙过大的表现明显不同,后者十分常见,且一般出生体重正常。③Silver-Russell 综合征(SRS):主要表现为颅面部异常(如相对巨颅、三角形脸、前额突出、 “鲨鱼嘴”、低耳位)、生后矮小、躯体不对称、小指侧弯等。SRS与KBGS在生后语言发育迟缓、智力低下、上颌中切牙多大的表现明显不同,后者较前者常见。
KBGS尚无特效的治疗方法,从减轻症状出发应该由遗传、神经、内分泌、骨科、物理康复治疗师组成多学科团队综合管理。矮小是该病的常见症状,但目前仅有1篇文献报道2例患儿用重组生长激素(GH)治疗,用药1年后身高分别增长0.6SDS及1SDS[29],治疗过程中未出现不良反应,表明GH治疗可能有效。本例患儿首次确诊矮小后即使用GH治疗用药14个月后,身高增长5.0 cm,提示GH治疗效果远低于生长激素缺乏的患儿,但可以帮助生长速度达到正常。
KBGS 属罕见遗传病,人类孟德尔遗传在线数据库(OMIM)表述为常染色体显性遗传,但亦有垂直传递的报道[10],提示其遗传方式仍待明确。完善本例患儿及文献复习分析提示,语言发育迟缓、学习障碍、智力低下、上颌中切牙过大、矮小及小指侧弯是本病核心表现,需进一步通过ANKRD11基因突变或(和)拷贝数变异检测明确诊断。
