苯丙酮尿症质谱筛查和基因诊断分析
2019-01-18陈梅刘清波江宁王伟红刘静黄萍孙茂
陈梅 ,刘清波 ,江宁 ,王伟红 ,刘静 ,黄萍 ,孙茂
1.唐山工人医院妇产科,河北唐山 063000;2.空军军医大学基础医学院,陕西西安 710032
苯丙酮尿症(Phenylketonuria,PKU,OMIM261600)是遗传代谢病中常见的常染色体隐性遗传病,上世纪30年代因佛伦医生(Folling)发现患者尿液中含有大量的苯丙酮酸而得名[1]。经过科研人员的研究苯丙酮尿症的发病机制已经很明确,其中≥90%是由于苯丙氨酸羟化酶(Phenylalaine hydorxylase,PAH)基因突变导致肝脏内酶活性降低或缺乏,≤10%是由于PAH的的辅酶四氢生物蝶呤 (Tetrahydrobiopterin,BH4)缺乏,两种情况都会造成苯丙氨酸(Phenylalanine,Phe)无法正常转化为酪氨酸(Tyrosine,Tyr),从而开启旁路代谢,产生大量的苯丙酮酸,并在体内异常蓄积,导致疾病发生。苯丙酮尿症其发病率因人群和地域而异,我国发病率约为1/11307[2]。如延误诊治,会造成患者神经系统不可逆损害,严重影响神经系统的发育和儿童的智力发育。该文回顾性分析2016年1月—2017年4月,通过该院DNA分型中心筛查的3 593例高危儿中确诊的8例苯丙酮尿症患儿情况,并结合分析了国外和国内的相关文献进行系统分析,以期加强临床医生对苯丙酮尿症的认识,现报道如下。
1 资料与方法
1.1 一般资料
该次检测在3 593例高危儿中检出8例PKU患儿,男3例,女5例,年龄1岁以下6例,8~10岁2例。在新生儿苯丙酮尿症和先天性甲状腺功能减低症筛查中得到PKU异常,进一步通过遗传代谢筛查(血液氨基酸和酰基肉碱检测、尿液有机酸检测)和基因诊断确诊苯丙酮尿症。该研究经医院医学伦理委员会通过,筛查人群(或监护人)都签署知情同意书。
1.2 研究方法
①液相色谱-质谱联用(LC-MS/MS)进行血液氨基酸和酰基肉碱检测。
②气相色谱-质谱(GC-MS)进行尿液有机酸检测。
③基因诊断。基因组DNA的提取;PCR扩增,PAH基因的引物序列参照文献设计[3],反应总体积为25 μL,循环条件为97℃ 5 min预变性,共35个循环(包括 94℃45s,55℃45s,72℃45s),72 ℃延伸 10 min,4℃保存。PCR扩增产物用1.5%的琼脂糖凝胶电泳检测后进行测序。
2 结果
2.1 代谢筛查结果分析
8例患儿遗传代谢筛查结果见表1,其中2例在血液氨基酸、酰基肉碱检测和尿液有机酸检测中都检出了相关物质,分别是患儿4:Phe为364.996 μM,尿液中检出苯乙酸 (Phenylacetic acid); 患儿5:Phe为954.070 μM,尿液中检测出苯乳酸(Phenyl lactate)和苯乙酸;6例在血液氨基酸和肉碱检测中Phe不同程度的上升,而尿液有机酸检测未见相关物质,3例未知。

表1 遗传代谢筛查结果
2.2 基因检测结果分析
8例患儿基因检测结果见表2,PAH共检测出7种突变类型,分别是内含子4上的剪接突变c.442-1G>A, 外显子 7 上的错义突变 c.721C>T、c.728G>A和终止突变c.781C>T,外显子12上的错义突变c.1238G>C、c.1256A>G、c.1301G>A。所有基因检测都对其父母进行了检测,证实了共分离。
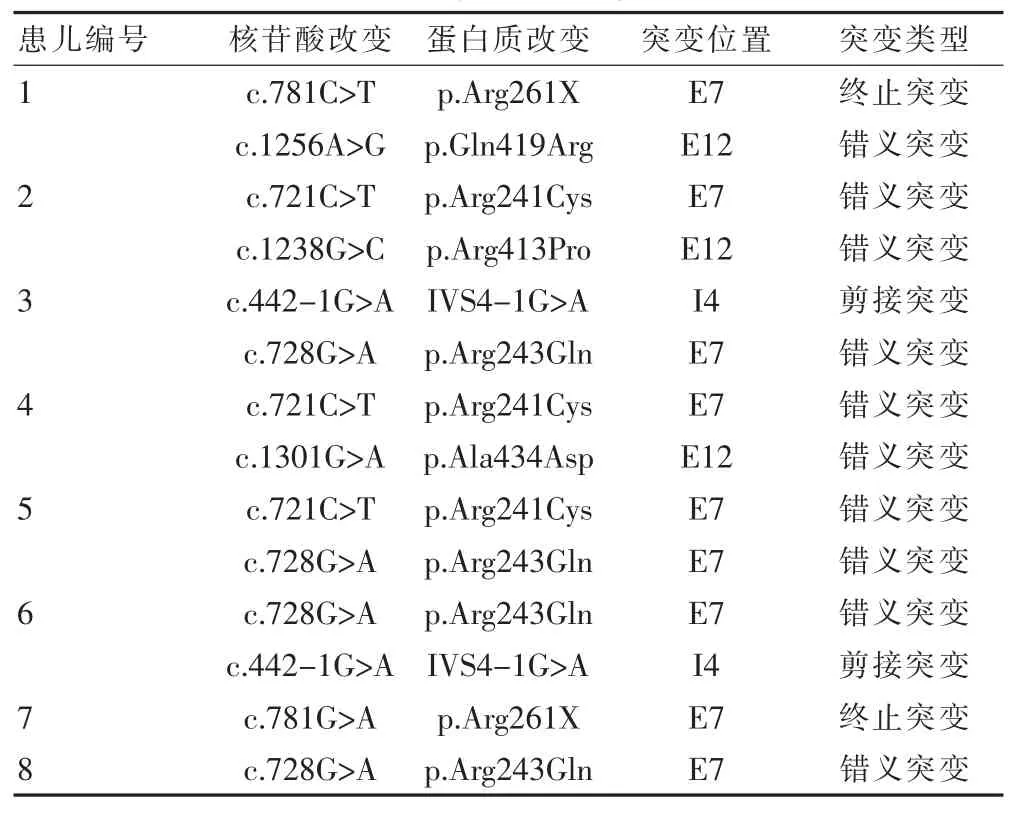
表2 代谢筛查和基因诊断结果
3 讨论
苯丙酮尿症其发病机制见图1,当苯丙氨酸不能转化为酪氨酸在体内异常蓄积,产生对人体有损害的苯丙酮酸和一系列旁路代谢产物,如苯乙酸、苯乳酸(患儿特征性的鼠尿味就是旁路代谢产物中的苯乳酸),会引起一系列神经系统损害。由于苯丙氨酸羟化酶只在肝脏细胞中表达,而在羊水和绒毛细胞中不表达,所以新生儿遗传代谢病筛查和基因诊断是实现疾病早期诊断的的有效手段[4]。通过研究PAH基因的突变谱,可以有效探究PAH基因型和临床表型的相关联性,也为研究苯丙酮尿症的遗传咨询和产前诊断打下了基础。
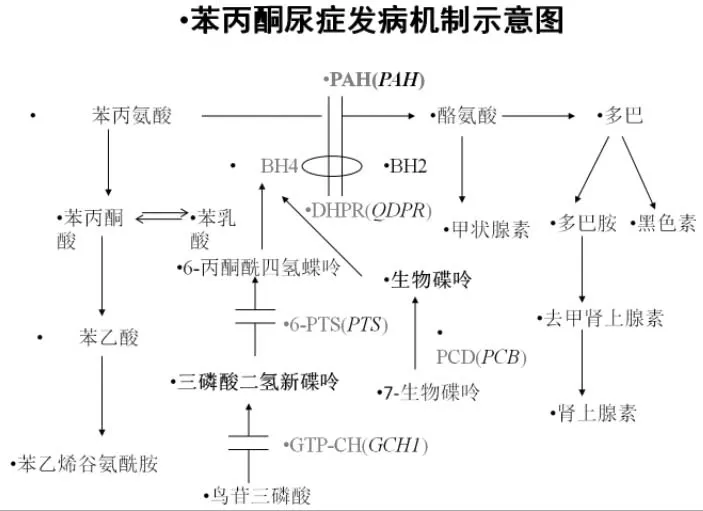
图1 苯丙氨酸羟化酶的代谢图
1983年科学家首次分离出了人类PAH基因,这为苯丙酮尿症的基因诊断奠定了基础。PAH基因位于12号染色体的长臂q22-q24.1,全长约100 kb,编码区包含13个外显子。迄今为止科学家已经发现一千余种突变,85%位于编码区的外显子上,15%位于内含子区、3’和5’区;根据对PAH酶活性有无影响,基因突变分为沉默突变和致病性突变。沉默突变顾名思义是对PAH酶活性无影响,而致病性突变多位于外显子、外显子和内含子的交接部分,突变直接影响PAH基因的转录、mRNA的翻译以及蛋白质的折叠与聚合的异常,进而影响酶的催化活性。
该研究共检测出7种突变类型,分别是内含子4上 c.442-1G>A, 外显子 7 上的 c.721C>T、c.728G>A和 c.781C>T,外显子 12上的 c.1238G>C、c.1256A>G、c.1301G>A,突变种类最多的集中在第7和12外显子,这与世界范围内突变最多的是PAH基因第7外显子,集中了大约16%的突变是一致的;因为第7外显子是编码PAH蛋白核心功能区,对保持该酶活性起关键性作用。
该研究突变次数最多的是c.728G>A共突变4次,该突变位点在中国人群中是高频突变位点,占总突变的18%[5],东北人中的频率18%,山西接近19%,南方人群略低9.5%,该位点已被证实为致病性突变[6]。东欧最常见的为第12外显子的c.1223G>A(R408W),俄罗斯远东地区占63%;第5外显子的c.473G>A(R158Q)基因突变在欧洲及拉美等国极为普遍,如荷兰的发生率为13.0%、比利时为9.0%、波兰、匈牙利、德国和捷克等国发生率都在5.0%以上[7],但该突变在中国突变频率却较低,只有0.8%。美国最常见的突变位点是 c.1241A>G(Y414C),约占 18.7%,这些说明PAH基因突变呈现高度种族和地区差异,热点突变在不同种族、不同地区均有不同。
亚洲PAH基因突变与欧美国家存在很大差异,日本苯丙酮尿症的发病率为1/78 400,最高频的突变是 c.1238G>C(R413P),占 30.5%;韩国苯丙酮尿症的发病率为 1/41 000,最高频的突变是 c.728G>A(R243Q),占12%[8];台湾地区苯丙酮尿症的发病率为1/55077,最高频的突变是 c.721C>T(R241C),占 32%[8]。
苯丙酮尿症患儿由于在新生儿期缺乏明显的特异症,这就给临床医生的早期诊治带来了困难,1953年德国Bickel医生首先应用了低Phe饮食治疗法获得成功,迄今该疗法仍然是治疗PKU最有效的方法。低Phe饮食疗法的治疗原则是既要使Phe的摄入量保证患儿生长发育和体内代谢的最低需要,又不使血中Phe过高。治疗过程中应给予特殊的低Phe奶粉,再配合其他低蛋白质食物和少量乳类,以达到最佳效果。值得注意的一点是,由于母乳中Phe的含量远远比其他食物要低,所以适当的搭配母乳喂养,也是有助于苯丙酮尿症患儿生长和发育的。近年也有研究学者不断提出新的治疗策略:如PKU特殊奶粉中加入L-肉碱;大分子中性氨基酸 (large neutral amino acids,LNAAs)的补充降低血 Phe浓度等[9-10]。 治疗过程中定期的监控是不可避免的,血液中Phe浓度检测是患儿近期饮食治疗控制情况的重要客观指标,不断的根据患儿的临床表现和各项检测指标,随时修改患儿的治疗方案,同时严密监测患儿体格、生长发育以及智力能力水平,进行有效的干预指导。
