同源DNA序列中间隔位点的核苷酸最近邻插补
2018-10-11秦雪瑞刘雄恩
秦雪瑞, 刘雄恩
(福建农林大学计算机与信息学院,福建 福州 350002)
分子系统发育分析是生物信息计算的一个重要分支,推算分子系统发育树可以重建祖先序列和估计分歧时间.通过分子系统发育研究可以探索生命的起源和物种间的进化历史,开展分类与区系研究以及流行病学、微生物生态学等的研究[1].
分子系统进化研究的第一步是建立同源性假设.DNA多序列比对数据代表了最初的同源性假设[2].一般情况下,参与比对的序列长度不是完全相同的,为了对齐需要插入间隔.比对序列的1列为1个位点,至少含有1个间隔的位点称为间隔位点,由共同祖先分歧后发生的插入或缺失事件引起.由于间隔起源于这种特殊的突变事件,包含适合于系统发育分析的历史信息[3],因此在分子系统发育分析中融合间隔位点的信息是有必要的.
常用的DNA进化马尔可夫模型,如JC69、K80、F81、F84、HKY85、TN93、REV94等,都只描述了4种核苷酸的置换(substitution)过程(本文统称这类DNA进化模型为4-状态模型),忽略了插入/缺失事件,在分子系统发育分析中应用这类模型势必会低估同源序列间的进化距离.将比对间隔视为碱基的第5种状态,Mcguire et al[4]首次提出了包含间隔位点信息的JC69+gap、F81+gap、F84+gap等模型,但这3种改进模型将插入/缺失事件与碱基置换或颠换(transversion)同等对待.2015年林碧娇等[5]在上述改进模型基础上引入新的参数,进一步区分了插入/缺失与碱基置换在性质上的差异,提出JC69+gap′、 F81+gap′、F84+gap′等模型,改进后的5种状态模型的参数较多,计算复杂,且仅在系统发育重建方法中的最大似然法上评估了应用效果.对于以上融合间隔位点信息的DNA进化马尔可夫模型(统称为5-状态模型),未在距离计算偏差上进行过有效分析.
为了在分子系统发育分析中尽可能多地融合indel信息,本文将多序列比对后出现的间隔视为统计抽样过程中产生的随机缺失数据.尝试以比对多序列的p距离矩阵表示序列间亲缘关系,依据最近邻原则选择碱基插补于特定序列的特定间隔位点,并比较分析插补前与插补后序列基于4-状态模型及插补前序列基于5-状态模型的序列间进化距离的大小,进而评估核苷酸最近邻插补法的有效性.
1 研究方法
1.1 最近邻插补的一般方法
最近邻插补根据研究对象在辅助变量上的接近程度来选择赋值单元,即利用辅助变量定义一个衡量单元间距离的函数,在无回答单元临近的回答单元中,选择与无回答单元距离最接近的回答单元所对应的值插补无回答值[6].其中,距离函数可根据应用实际采用不同的距离测度.
对于离散化矩阵,最近邻插补法一般采用匹配度来计算样本单元i和j之间的距离[7].记m为样本单元个数,n为属性类数,则样本间的距离为:
(1)
式中,i,j= 1,2,…,m.Ai为含缺失数据的样本单元i的属性向量;Aj为其他各样本单元j的属性向量;ail为样本单元i在属性l上的值.要求属性值向量Ai和Aj在属性l上无缺失值.
(2)
可见,dij为第i个和第j个样本单元的属性值向量Ai和Aj中属性不同的属性个数.属性个数越少,两样本单元距离越近.
设样本单元i的属性t缺失,则插补函数为
(3)
样本单元i与k距离最小且k在t上的属性值akt存在,则样本单元i在t上的属性值ait插补为akt.
由于最近邻插补算法计算简便,效果明显,在缺失数据处理中都有着广泛的应用.
1.2 同源DNA序列中间隔位点的核苷酸插补
将同源DNA比对序列视为统计抽样的多个样本单元,每个位点独立进化,位点视为样本单元的属性,比对序列中的间隔即为缺失数据.在分子系统发育分析中,由于针对比对后的同源序列间无论使用观察距离(即p距离[8])还是基于核苷酸替代模型的进化距离,反映的物种间亲缘关系远近的顺序是一致的,而p距离通过2个序列中非同一核苷酸位点的比例来测度分歧大小,即
(4)
式中,mij和nij分别为序列i与序列j中非同一核苷酸位点数和位点总数.
基于最小进化原理[9],以序列间p距离中最短距离作为最近邻的依据,间隔位点核苷酸插补函数为:
(5)
即,序列Si与Sk的p距离最短且Sk在位点t上存在核苷酸Skt,则Sit的间隔插补为Skt.
同源DNA序列中间隔位点核苷酸最近邻插补算法描述如下:
Algorithm Nucleotide Interpolation by NNI
Begin
Input multi-aligned DNA sequencesS
Computingp-distance matrixP
Fort← 1 st To the last gap site Do
Begin
Fori← 1 st To the last sequence with gap attDo
Ifpik=minj(pij) andSkt∈{A,T,C,G} ThenSit←Skt
Forj← 2 nd To the last sequence Do
IfSjt≠SitThen break Else continue loop
Ifj> count of sequences Then delete sitetElse remaint
End
OutputSafter Nucleotide Interpolation at gap sites
End
核苷酸最近邻插补算法:先计算p距离矩阵;然后针对多序列比对的核苷酸矩阵,对含有间隔的所有列中的每个存在间隔的序列,选择与该序列距离最近且在该位点没有间隔的核苷酸,将其在该位点的间隔进行替换,即插补.若插补后各序列在该位点的核苷酸完全相同,则删除该位点(整列),否则保留插补后位点.剔除插补后核苷酸相同的位点,是因为原来的间隔位点代表可能的indel突变事件,而插补后这种位点在分子进化分析中不提供进化信息,反而会减低序列间进化距离的估算.
假设在p距离上,与序列①最近的是序列②,与序列②最近的是序列①,与序列③最近的是序列②,与序列④最近的是序列⑤,与序列⑤最近的是序列④.绿色线框标注的是最近邻插补后核苷酸不同的位点,红色线框标注的是插补后核苷酸相同而删除的位点.

图1 核苷酸最近邻法插补示意图Fig.1 Schematic diagram of nucleotide interpolation by nearest neighbor method
1.3 几种DNA进化模型下的进化距离
DNA进化的马尔可夫模型以不同状态(4种核苷酸,或再加上1个gap状态)间置换的速率进行矩阵描述.基于这类模型可以推导出序列间进化距离(平均每个位点核苷酸置换次数)的计算公式[8].为便于测试和比较本文提出的核苷酸最近邻插补处理方法与传统的直接忽略间隔位点的方法,以及将间隔视为第5种状态的改进模型的方法,本文采用F81、F84、F81+gap、F84+gap和F81+gap′等模型下的距离.
1.3.1 F81模型 Felsenstein[10]在JC69模型置换速率矩阵中引入4个核苷酸的比例(平衡频率),提出F81模型.当同源序列中4种核苷酸的比例存在偏倚,且转换和颠换位点比例均衡时F81模型较为有效.F81模型下导出的距离为:
(6)
式中,a=2(πTπC+πAπG+πYπR),πR=πA+πG,πY=πT+πC,πT、πC、πA和πG分别为4种核苷酸T、C、A和G的平衡频率,计算时以观察值估算;p为距离.
1.3.2 F84模型 Felsenstein et al[11]将核苷酸置换区分为两类事件,类型Ⅰ仅含有转换,类型Ⅱ既有转换又有颠换,同时两种类型里都有1个核苷酸都可以被相同的核苷酸置换,即核苷酸不发生改变.F84模型能较好地拟合进化过程中核苷酸的变化情况[5].该模型导出的距离为:
(7)

式中,S是转换位点的概率,V是颠换位点的概率.显然有p=S+V.
1.3.3 F81+gap模型 Mcguire et al[4]在F81模型中引入第5种状态,即比对间隔,其与4种核苷酸的置换采用核苷酸之间的置换速率,该模型导出的距离,表示如下:
(8)
式中,a=2[πTπC+πAπG+πYπR+π_(1-π_)],π_是间隔的平衡频率.
1.3.4 F84+gap模型 Mcguire et al[4]在F84模型中同样引入间隔状态,将核苷酸转换用速率α表示,而嘧啶和嘌呤的之间的颠换、4种核苷酸与间隔的置换用另一速率β表示.F84+gap模型导出的距离为:
(9)

1.3.5 F81+gap′模型 在考虑核苷酸平衡频率因素的同时,将核苷酸之间的置换与核苷酸和间隔之间的置换(即插入/缺失)区别对待,在F81+gap′模型中,引入参数γ表示核苷酸与间隔间的置换速率.该模型导出的距离为:
(10)
式中,a=2(πTπC+πAπG+πYπR),b=1/[a+2π_(1-π_)],S是核苷酸置换位点的概率,I是核苷酸与间隔间置换位点的概率.显然有,p=S+I.
2 结果与分析
2.1 测试序列
分别选取3组同源DNA序列进行测试.第1组为7种猿类物种的线粒体DNA全序列,物种及其序列GenBank检索号分别为Pantroglodytes(NC_001643.1)、Panpaniscus(NC_001644.1)、Homosapiens(NC_012920.1)、Pongopygmaeus(NC_001646.1)、Pongoabelii(NC_002083.1)、Gorillagorilla(NC_001645.1)、Hylobateslar(NC_002082.1).用ClustalX2默认的参数进行多比对、手工优化后,序列长度为16 644 bp,其中间隔位点为419 bp,数据缺失率为2.5%.
第2组为6属6种睡莲科植物的核糖体DNA中的内转录间隔区(ITS)序列[12],分别为Nelumbopentapetala(AY620419.1)、Nymphaeacaerulea(AY620420.1)、Victoriacruziana(AY620423.1)、Cabombafurcata(AY620425.1)、Braseniaschreberi(AY620426.1)、Nupharlutea(AY620427.1).比对后序列长度为673 bp,其中间隔位点181 bp,数据缺失率为26.9%.
第3组为真菌侧耳属8个种的25S rDNA序列,分别为Pleurotusabieticola(AF135176.1)、Pleurotusaustralis(AF261432.1)、Pleurotuscalyptratus(AF135177.1)、Pleurotuscornucopiae(U04146.1)、Pleurotusdryinus(AF135178.1)、Pleurotusfossulatus(U04136.1)、Pleurotuspopulinus(U04159.1)、Pleurotussmithii(U04150.1).比对后序列长度为903 bp,其中间隔位点53 bp,数据缺失率为5.9%.
2.2 测试结果
表1显示第1组数据分别在删除间隔位点后4-状态模型、融合间隔位点5-状态模型和间隔插补核苷酸后4-状态模型下的成对序列间进化距离.
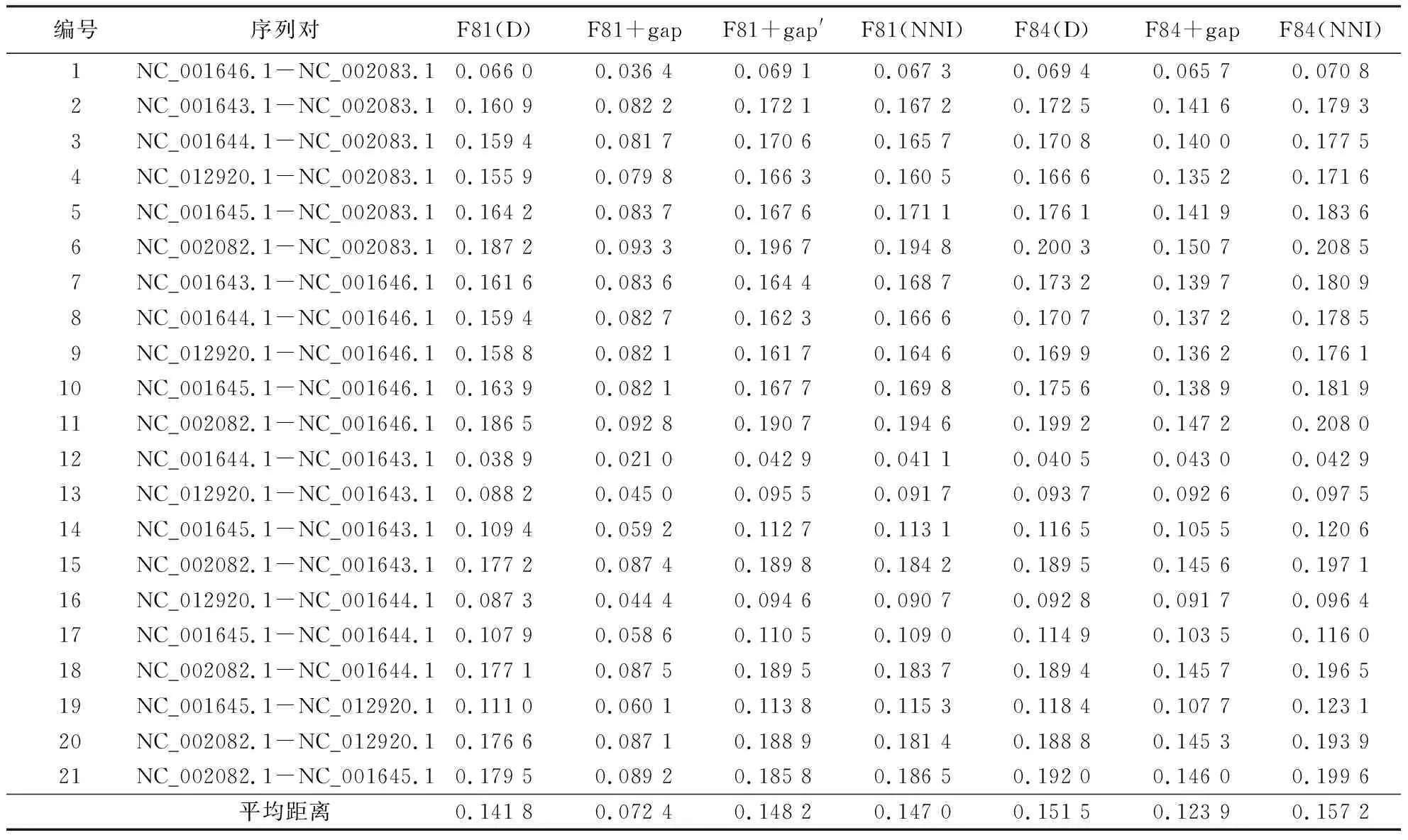
表1 猿类7个物种线粒体DNA序列在几种处理和模型下成对进化距离1)Table 1 Evolutionary distances of mitochondrial DNA sequences of 7 apes under several processings and models
1)F81(D)为删除所有间隔位点后采用F81模型的距离,F81(NNI)为最近邻法核苷酸插补间隔后采用F81模型的距离,F84(D)为删除所有间隔位点后采用F84模型的距离,F84(NNI)为最近邻法核苷酸插补间隔后采用F84模型的距离.
表2显示第2组数据分别在删除间隔位点后4-状态模型、融合间隔位点5-状态模型和间隔插补核苷酸后4-状态模型下的成对序列间进化距离.
表3显示第3组数据分别在删除间隔位点后4-状态模型、融合间隔位点5-状态模型和间隔插补核苷酸后4-状态模型下的成对序列间进化距离.
表1~3中的序列间平均距离以及图2~4中针对间隔位点的不同处理或模型下估算距离的对照直观地表明:融合间隔位点信息的5-状态模型中的F81+gap和F84+gap的距离估算明显偏低,改进的5-状态模型F81+gap′、传统的删除间隔位点的处理和本文提出核苷酸最近邻插补处理后4-状态模型估算的距离相对接近,而改进的F81+gap′模型和核苷酸最近邻插补处理后在4-状态模型下估算的距离略高于直接忽略间隔位点信息在4-状态模型下的估算,且核苷酸最近邻插补处理方法估算的距离又略高一些.其次,序列间间隔位点数越大,忽略间隔位点方法造成的进化距离偏低估计越加突出.

表2 睡莲科6种植物核糖体DNA中ITS序列的成对进化距离Table 2 Evolutionary distances of ITS sequences in ribosomal DNA of 6 Nymphaeaceae plants

表3 侧耳属8种真菌25S rDNA序列的成对进化距离Table 3 Evolutionary distances of 25S rDNA sequences of 8 Pleurotus fungus
由于间隔位点代表DNA突变中的核苷酸插入/缺失事件,直接删除同源多序列比对后的间隔位点的简单处理方法势必导致序列间进化距离的偏低估计,应用于分子系统发育分析和进化树推断时将低估序列间距离,造成枝长偏低估计.5-状态模型中的F81+gap和F84+gap更加低估了序列间距离,本文认为这是由于这两个模型均没有区分核苷酸之间的替代与核苷酸与间隔之间的置换(插入/缺失),简单地处理为相同性质、同一置换速率的状态转换过程.

图2 不同方法估算的7种猿类线粒体DNA序列间距离对照Fig.2 Comparison of estimated distances of mitochondrial DNA sequences of 7 apes under different methods

图3 不同方法估算的6种睡莲科植物核糖体DNA中ITS序列间距离对照Fig.3 Comparison of estimated distances of ITS sequences in ribosomal DNA of 6 Nymphaeaceae plants
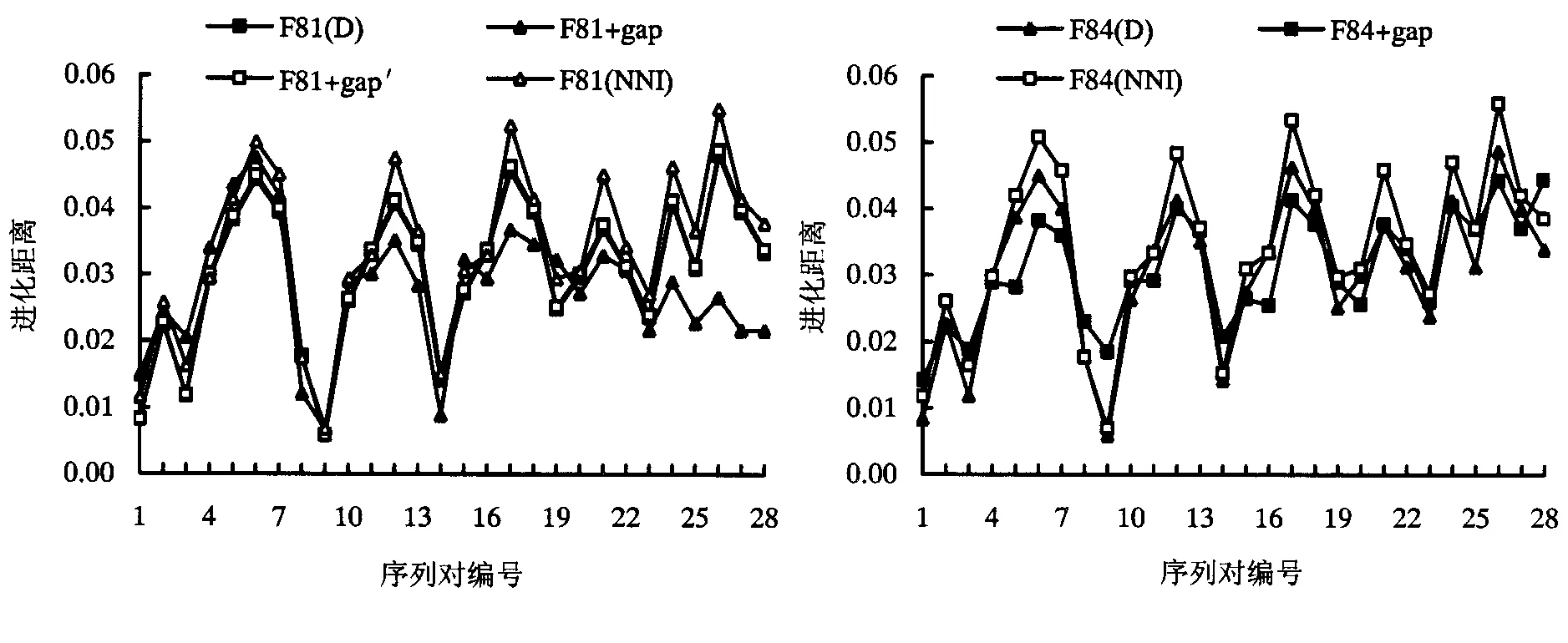
图4 不同方法估算的8种侧耳属真菌25S rDNA序列间距离对照Fig.4 Comparison of estimated distances of 25S rDNA sequences of 8 Pleurotus fungus under different methods
睡莲科6种植物核糖体DNA中ITS序列的成对进化距离的测试结果(表2和图2)表明,当序列间隔位点数较多,即DNA进化过程中核苷酸插入/缺失事件的比例较高时,本文提出的最近邻核苷酸插补方法在进化距离和进化树枝长估算上能更为有效地消除偏低估计,对间隔位点进行核苷酸插补的处理方法使得传统的4-状态模型在序列间分歧度的估算中能够更有效地融合DNA进化的插入/缺失信息.
改进的F81+gap′模型和核苷酸最近邻插补处理方法至少能够减少同源序列间距离的偏低估计.如果核苷酸最近邻插补的方法没有导致进化距离的偏高估计,无疑是一种有效的融合InDel信息的方法.
3 小结
鉴于分子系统发育重建研究中忽略多序列比对出现的间隔位点而导致低估序列间进化距离或进化树枝长的问题,本文借鉴统计学中处理缺失数据的最近邻插补法,提出一种核苷酸最近邻插补间隔位点的处理方法.通过对3组同源DNA序列在不同的处理方法下的距离估算对照测试和上述分析,本文发现将间隔视为4种核苷酸外的第5种状态的F81+gap和F84+gap模型不能有效融合间隔所表示的indel进化信息,反而更加低估了序列间距离,改进的同类模型F81+gap′能够在一定程度上融合间隔所携带的indel信息,而本文所提出的核苷酸最近邻插补法能够有效运用DNA进化的4-状态马尔可夫模型估算进化距离,至少它能减小序列间进化距离的偏低估计,至于是否出现偏高估计还需要通过对DNA模拟进化序列进行分子系统发育重建和分析做出进一步判断.
