高效液相色谱法检测水产品中三种喹诺酮类药物残留量方法研究
2018-09-05童文羽朱志强
童文羽 朱志强 曾 智
本试验改进了农业部783号公告-2-2006水产品中诺氟沙星、环丙沙星、恩诺沙星残留量的高效液相色谱法,并对样品的提取条件和色谱方法进行了优化分析,确定使用盐酸酸化乙腈提取,固相萃取小柱净化,高效液相色谱-荧光检测器检测、外标法定量的分析方法。本方法漂移率小,对三种喹诺酮类药物标准曲线回归系数在0.999以上,线性范围1~1000μg/L,定量限1μg/kg。对草鱼、对虾样品进行三种喹诺酮类药物加标回收率的验证实验,回收率在79.2%~105.0%之间,相对标准偏差在0.83%~9.85%之间,批次间标准偏差在5.00%~10.57%之间,说明方法前处理提取效率和准确度满足分析要求。
1.材料与方法
1.1 仪器、试剂、标准品与耗材
试验材料来自历次农业部例行采集样品,品种有草鱼和对虾。
岛津LC-20AD高效液相色谱仪(配荧光检测器RF-20A),LC Solution工作站与Waters e2695高效液相色谱仪(配荧光检测器2475),Empower 3工作站;色谱柱为Thermo Acclaim C18与安捷伦ZORBAX SBC18,规格均为4.6mm×250mm,I.D.5μm。
三种沙星标准品均为Dr.Ehrenstorfer。其中诺氟沙星纯度为99.1%,环丙沙星纯度为95.0%,恩诺沙星纯度为99.0%。分别称取诺氟沙星、环丙沙星、恩诺沙星标准品各0.01g(精确至0.0001g),用色谱纯级甲醇溶解,必要的时候使用超声助融,定容到100mL棕色容量瓶中,4℃冷藏保存,有效期6个月。
试验使用试剂及其他仪器规格如下(表1-表3):
1.2 实验前处理的优化
1.2.1 样品的处理方法
取鱼、虾肌肉,切成小块,匀浆机打成匀浆后准确称取5.00g(±0.05g)样品,置于50mL离心管中,加入15mL提取剂,盖紧盖子,置于振荡器上振荡5min,4500r/min离心5min,上清液倒入另外一支50mL离心管中。
表1 试验使用的各试剂品牌个规格
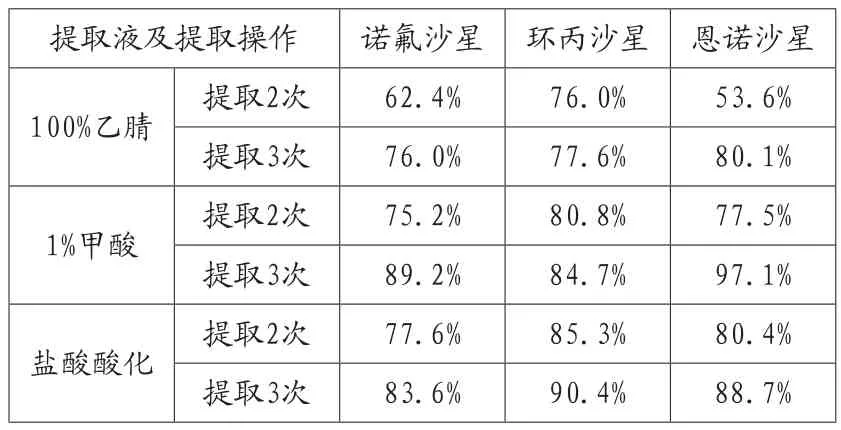
表4 甲酸酸化和盐酸酸化的乙腈提取的回收率

表5 提取次数的回收率
1.2.2 提取液的选择和优化
根据农业部783、1077号公告,选择酸化乙腈作为提取剂,因为喹诺酮类都是两性化合物,提取时在提取剂中加入酸会提高提取效率。分组使用100%乙腈、1%甲酸酸化和每2500mL提取剂加入20mL50%盐酸进行酸化的乙腈作为提取剂,每次提取用量15mL。

表2 固相萃取小柱品牌、规格

表3 其他仪器规格型号
从表4可以看到,由于这三种喹诺酮类药物都是两性化合物,酸化的提取剂可以大大提高提取回收率,而提取实验也证明了这一点。其中盐酸酸化的乙腈作为提取剂的回收率最高,原因是盐酸的酸性较甲酸强。同时,提取3次比2次显著提高回收率。对提取次数进行进一步优化结果如表5,提取4次后,诺氟沙星回收率有提高,但环丙沙星和恩诺沙星的回收率并没有显著提高,因此采用盐酸酸化的乙腈作为提取剂,提取3次即可。
1.2.3 样品的净化方法的优化
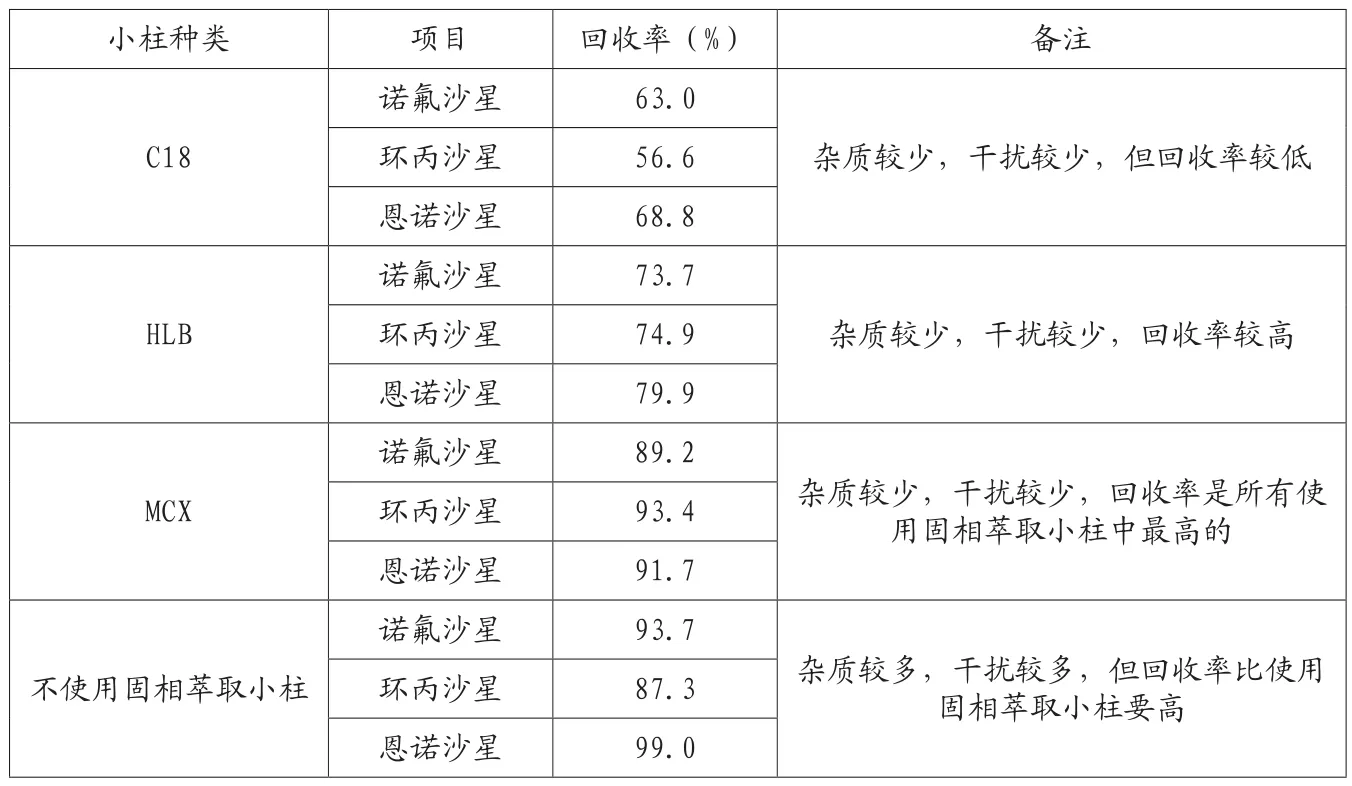
表6 3种固相萃取小柱以及不使用小柱的加标回收率
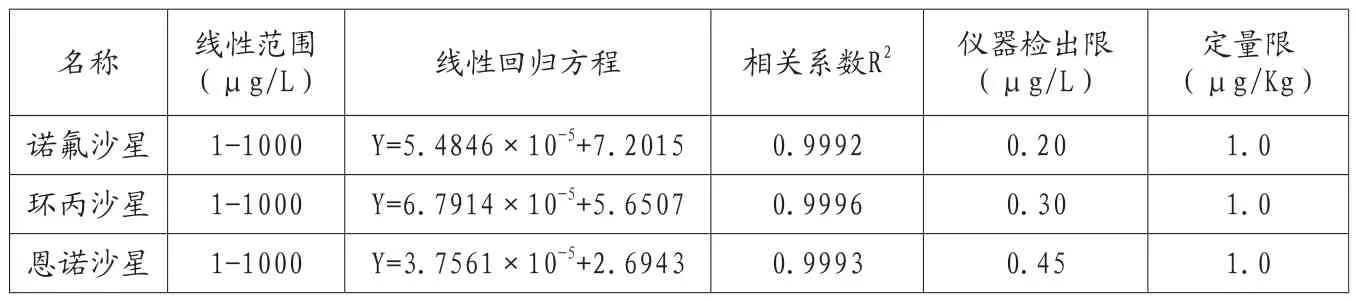
表7 方法的线性范围、线性回归方程、相关系数和检测限
喹诺酮类因为是两性化合物,所以在正己烷中的溶解度很差。因此正己烷在喹诺酮类的前处理过程中常用作去杂试剂。
旋转减压蒸馏或氮吹干提取液后,用2.0mL磷酸二氢钾-甲醇溶液溶解残渣(34.0g磷酸二氢钾溶于230mL水,用25%氢氧化钠调pH至7.0,用该溶液将20mL甲醇稀释至100mL),在提取液中加入2mL正己烷,涡旋,静置后去掉上层的正己烷,正己烷净化操作需要进行2次,如果样品基质特别复杂,净化次数可酌情增加。
根据唐巍、卢艳芬等人报道,水产品中喹诺酮类残留量测定前处理中可使用Waters Oasis HLB固相萃取小柱净化,农业部1025号公告则使用C18小柱,因此将包括这两种小柱在内的一共3种固相萃取小柱以及不使用固相萃取净化分组做对比实验。
所有的萃取小柱在使用前均使用3.0mL甲醇和3.0mL水激活萃取柱,C18、HLB、MCX、PRS小柱激活后加入备用样品过柱,6.0mL纯水淋洗去杂,用6mL的6%氨化甲醇(6mL氨水用甲醇稀释至100mL)洗脱,收集洗脱液,于45℃氮吹至干,1.0mL流动相溶解残渣,经滤膜过滤,供液相色谱分析使用;
不使用固相萃取小柱净化的对照组,在第一次氮吹后直接使用1mL流动相溶解残渣,定容后过滤供液相测定使用。

图1 流动相为乙腈:四丁基溴化铵-磷酸缓冲液体积比5∶95的标准品重叠图
根据表6的统计,不使用萃取小柱回收率是很高的,缺点是色谱图上的杂质依然较多,而使用固相萃取小柱后,尽管回收率都出现了不同程度的降低,但从仪器图谱上来看,使用固相萃取小柱可以显著的减少杂质峰,减少干扰。因此使用萃取小柱是有意义的。
1.3 色谱条件
农业部783号公告中使用的是乙腈∶四丁基溴化铵-磷酸(pH3.1)=95∶5作为流动相,在实际实验过程中发现以下几个问题:
由于喹诺酮类属于两性分子,传统的色谱柱常常会因为残余硅醇基和金属不纯物的存在而导致色谱峰拖尾。在农业部783号公告的方法中,在流动相中加入四丁基溴化铵试剂(3.22g四丁基溴化铵溶于1000mL水中,磷酸调pH值为3.1),作为离子对试剂降低其极性,从而改善峰形和提高分离度。但喹诺酮类样品在色谱柱保留时间对pH 值很敏感,而离子对试剂本身无法起到有效的缓冲作用,可能造成目标物质保留时间的漂移,如图1。
可以看到乙腈-四丁基溴化铵流动相虽然峰型和分离度较好,但出现了在保留时间有漂移。同时流动相中的四丁基溴化铵很容易产生气泡,即便长时间超声脱气也很难改善,因此参考农业部1025号公告,使用乙腈-磷酸三乙胺(3.4mL浓磷酸溶于1000mL水中,三乙胺调pH值为2.4)作为流动相。其中乙腈:磷酸-三乙胺缓冲液体积比为18∶82,流速1.0mL/min;使用荧光检测器,激发波长280nm,发射波长450nm;柱温35℃;进样量20μL。该色谱条件下的标准品叠加如图2,可以看到目标峰的漂移相比乙腈-四丁基溴化铵作为流动相时改善了很多,而且出峰较快,节省了时间和流动相。
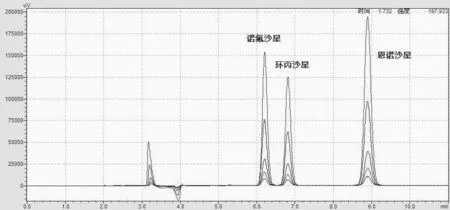
图2 流动相为乙腈∶磷酸-三乙胺缓冲液=18∶82的标准品重叠图

图3 3种喹诺酮类标准品色谱图(三种沙星均为5μg/L)
2.结果与讨论
2.1 色谱分离
在优化出的色谱条件下,分析得到标准溶液、空白和加标样品的色谱图,见图4。可以看到,在所使用的色谱条件下,目标峰得到了很好的分离。
2.2 检出限和定量限
取三种喹诺酮类标准储备液,用流动相稀释成标准工作液,在优化出的色谱条件下进样,以各组分质量浓度与色谱峰面积进行线性回归,绘制工作曲线,得到回归方程如表7。
因为实际工作中样品的基质较为复杂,因此,为减少杂峰干扰,故选用了10倍信噪比(S/N)计算仪器检出限,诺氟沙星、恩诺沙星、环丙沙星的仪器检出限为0.20μg/L、0.30μg/L、0.45μg/L。同时,根据加标实验情况以及同时测定的要求,确定三种沙星的定量限为1.0μg/Kg。

表8 方法的回收率
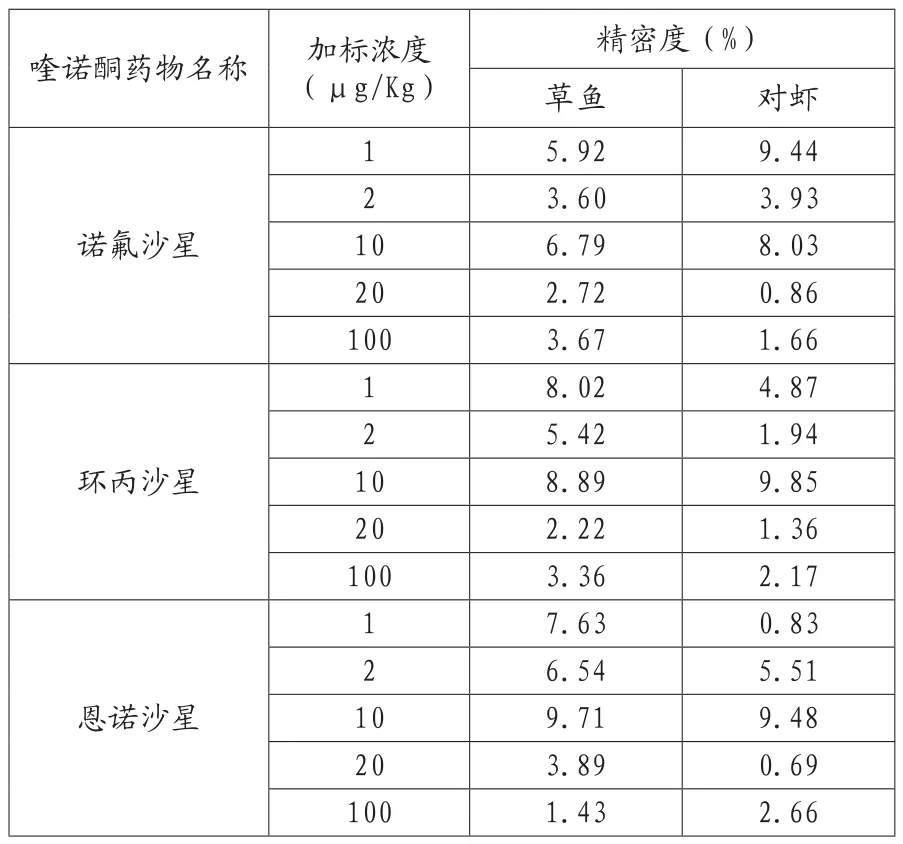
表9 方法的精密度
2.3 回收率和精密度
分别对草鱼和对虾样品加标,测定加标回收率和方法精密度。测定结果见表8-9,三种喹诺酮类在草鱼和对虾两种样品中的平均回收率在79.2%~105.0%之间,相对标准偏差在0.83%~9.85%之间,批次间相对标准偏差在5.00%~10.57%之间。
2.4 结论
本研究采用了盐酸酸化乙腈提取、正己烷和Oasis MCX固相萃取小柱净化,使用乙腈-磷酸三乙胺作为流动相,建立了水产品中恩诺沙星、诺氟沙星、环丙沙星残留量的高效液相色谱-荧光检测测定方法。该方法较之农业部783号、1025号公告方法有着定性、定量更加准确,精密度高,重现性好,能满足我国现行兽药残留检测分析要求,可用于水产品中诺氟沙星、环丙沙星、恩诺沙星药物残留量的测定。
