利用回归模型筛选出近天然的抗原-抗体对接模拟结构
2018-06-26陈郑珊迟象阳范鹏飞张冠英王美荣于长明陈薇
陈郑珊,迟象阳,范鹏飞,张冠英,王美荣,于长明,陈薇
军事科学院军事医学研究院 生物工程研究所,北京 100071
随着计算机性能不断增强以及模拟计算方法不断成熟,出现了应用在生物领域的分子模拟方法,能够通过计算机模拟对生物大分子进行研究。在众多的模拟方法中,分子对接已成为其中最重要和应用最广泛的方法之一。分子对接主要是考察和预测两个分子在复合物中的结合模式,分子对接在抗原-抗体的研究中有着重要的应用。在抗体-抗原性质的研究中,表位信息是研究人员最为关心的方面之一,相对于主流的实验方法,分子对接模拟在抗原表位 (尤其是空间构象性表位)的预测和辅助筛选方面具有特别的优势[1],分子对接结果能够提供对阐明抗体中和机制有价值的线索[2-3]。然而在对接产生的大量对接构象中,近天然结构占很小的比例,如何将这些结构筛选出来仍是一个具有挑战性的问题[4]。相关工作有所进展[5-6],但目前尚无一个普适性好、准确性高的打分函数可以实现这一筛选目的。本文将 QSAR的原理应用于抗原抗体对接模拟构象的筛选 (近天然结构预测)。即用数理统计方法抽提抗原-抗体复合物模拟构象的近天然程度与其抗原-抗体接触面的理化特性、能量特性之间的定量变化规则。通过对抗原-抗体接触面描述符和能量参数的回归分析,建立用于筛选近天然构象的数学模型。所得数学模型主要适用于抗原-抗体对接体系,在测试集验证和埃博拉病毒的包膜蛋白4G7抗体结合模式预测应用中具有较好的表现。
1 材料与方法
1.1 抗原-抗体接触面描述符和能量参数
候选的抗原抗体接触面描述符:①接触面面积;②接触面上氢键密度;③接触面上 cation-π密度;④EPII[7](Epitope-paratope interface index);⑤ZDock Score[8](基于格点的几何互补性打分);⑥ZRank Score[9];⑦ ZRank VdW;⑧ ZRank Elec;⑨ZRank Solv。其中,ZRank Score是ZRank VdW
(Van der Waals energies)、ZRank Elec (Electrostatics energies) 和 ZRank Solv (Desolvation energies) 的线性组合。EPII是抗原-抗体接触面上氨基酸对偏好系数的线性组合:
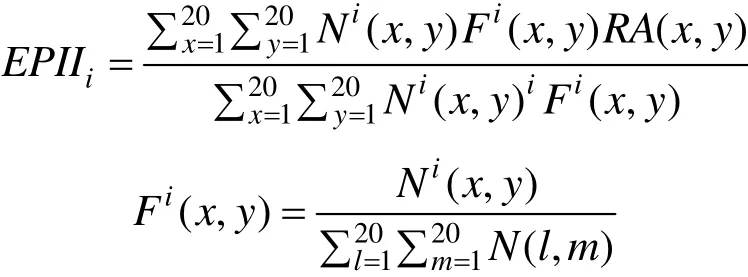
Ni(x,y)表示氨基酸对(x,y)在接触面i上的数量,Fi(x,y)则表示表示氨基酸对(x,y)在接触面i上出现的频率 (注:x表示抗原上的氨基酸,y表示抗体上的氨基酸)。Tharakaraman等[7]统计了84个抗原-抗体复合物结构接触面上的氨基酸对出现的频率,表示为20×20的矩阵RA,作为抗原-抗体接触面上氨基酸对偏好系数矩阵。接触面上残基类型的偏好性[10]可能与接触面上广泛存在的阳离子-π (Cation-π) 相互作用有关[11],将接触面上 cation-π密度列为候选的描述符。ZDock Score基于格点算法表征两个对接单体的形状互补性。ZRank Score表征范德华作用能、静电作用能和溶剂化作用能的综合影响。
接触面描述符及能量参数⑤⑥⑦⑧⑨由BIOVIA Discovery Studio 4.5软件平台的对接模拟程序 (ZDOCK[8]) 计算得到;①②③④是自行使用 Perl语言编写程序计算得到,BIOVIA Discovery Studio 4.5软件[12]的客户端所提供的应用程序编程接口 (Discovery Studio scripting API),相当于Perl语言的扩展函数库,为编写程序操作生物分子模型及相关数据的处理提供了很大的便利。
1.2 模拟结构准确程度标准的定义
将两个对接单体 (即抗原和抗体) 上距离另一个单体不超过4.5 Å的氨基酸定义为接触面氨基酸。将两个复合物结构中接触面上的相同氨基酸重叠后,计算接触面氨基酸上的重原子 (非氢原子) 的RMSD值,即I_RMSD[13]。I_RMSD描述了抗原-抗体接触面在原子水平上的准确度。如果某个模拟对接结构与相应实验测定的晶体结构 (下载自PDB 数据库http://www.rcsb.org/) 的I_RMSD小于2.0 Å,则认为该模拟对接结构是近天然结构。
1.3 抗原-抗体分子对接
在BIOVIA Discovery Studio 4.5生命科学分子模拟软件平台上使用 ZDOCK[8]程序进行分子对接得到抗原-抗体反应的计算生成构象模型。对接过程主要使用抗体的可变区部分作为受体,使用抗原作为配体。ZDOCK计算过程中采用6°欧拉角度进行结合构型采样 (构象空间搜索),最终样本包括54 000个结合构象模型。
1.4 准备训练集和测试集数据
选用由 Hwang等[14]提出的 Protein-Protein Docking Benchmark Version 4.0中的抗原-抗体复合物结构作为筛选算法的研究对象。所选用的 37个抗原-抗体复合物结构均收集自PDB (Protein data bank)[15],分辨率高于3.25 Å,氨基酸链长度不少于30个残基,抗体部分均包含轻链和重链可变区。在全部 37个抗原-抗体复合物结构中再随机选取19个抗原-抗体复合物结构 (表 1) 用于数学模型的训练,其余18个抗原抗体复合物结构 (表2) 作为测试对象。对于每一个的抗原-抗体复合物结构,其本身是实验测定结构,作为标准结构使用,拆分出其两个单体 (即抗原和抗体) 进行ZDOCK对接得到54 000个对接模拟构象 (Decoy model),分别计算出每个对接模拟构象与标准结构间的I_RMSD值。从ZDock Score打分排序在前2 000名的对接模拟构象中选取I_RMSD值最小者 (要求其I_RMSD值必须小于2.0 Å) 作为近天然结构,如果该 2 000个对接模拟构象的I_RMSD值均不小于2.0 Å,则以相同标准从全部54 000个对接模拟构象中选取 I_RMSD值最小者。再对该2 000个对接模拟构象进行聚类分析,RMSD Cutoff参数设定为10.0,即同一个聚类簇(Cluster) 中的不同构象模型间的RMSD值<10 Å,而来自不同簇间的构象模型的RMSD值≥10 Å。聚类分析得到101个聚类簇,除去近天然结构所在的聚类簇后 (如果近天然结构不属于任何一个聚类簇,则除去包含对接模拟构象最少的一个聚类簇),选取余下100个聚类簇的代表元作为不合理结构。即从对接结果中选出 1个近天然结构(I_RMSD<2.0 Å) 和 100个不合理结构,并构成一个具有101个构象元素的训练集体系或测试集体系。总共得到19个训练集体系和18个测试集体系。需要说明的是,其中有1个训练集体系由101个不合理构象组成 (因为54 000个对接模拟构象中的I_RMSD值均不小于2.0 Å)。
通过BIOVIA Discovery Studio 4.5软件和自行设计编写的Perl语言程序的计算得出训练集体系和测试集体系中所有对接模拟构象的接触面描述符和能量参数。在各体系内对各项参数进行标准化(归一化) 处理,作为数学模型的自变量。定义因变量Y,对于训练集体系中的每一个对接模拟构象,若该构象是近天然结构,则Y=1;若该构象是不合理结构,则Y=0。其中数据的批量处理和格式转换等操作也是由自行编写的Perl语言程序完成。
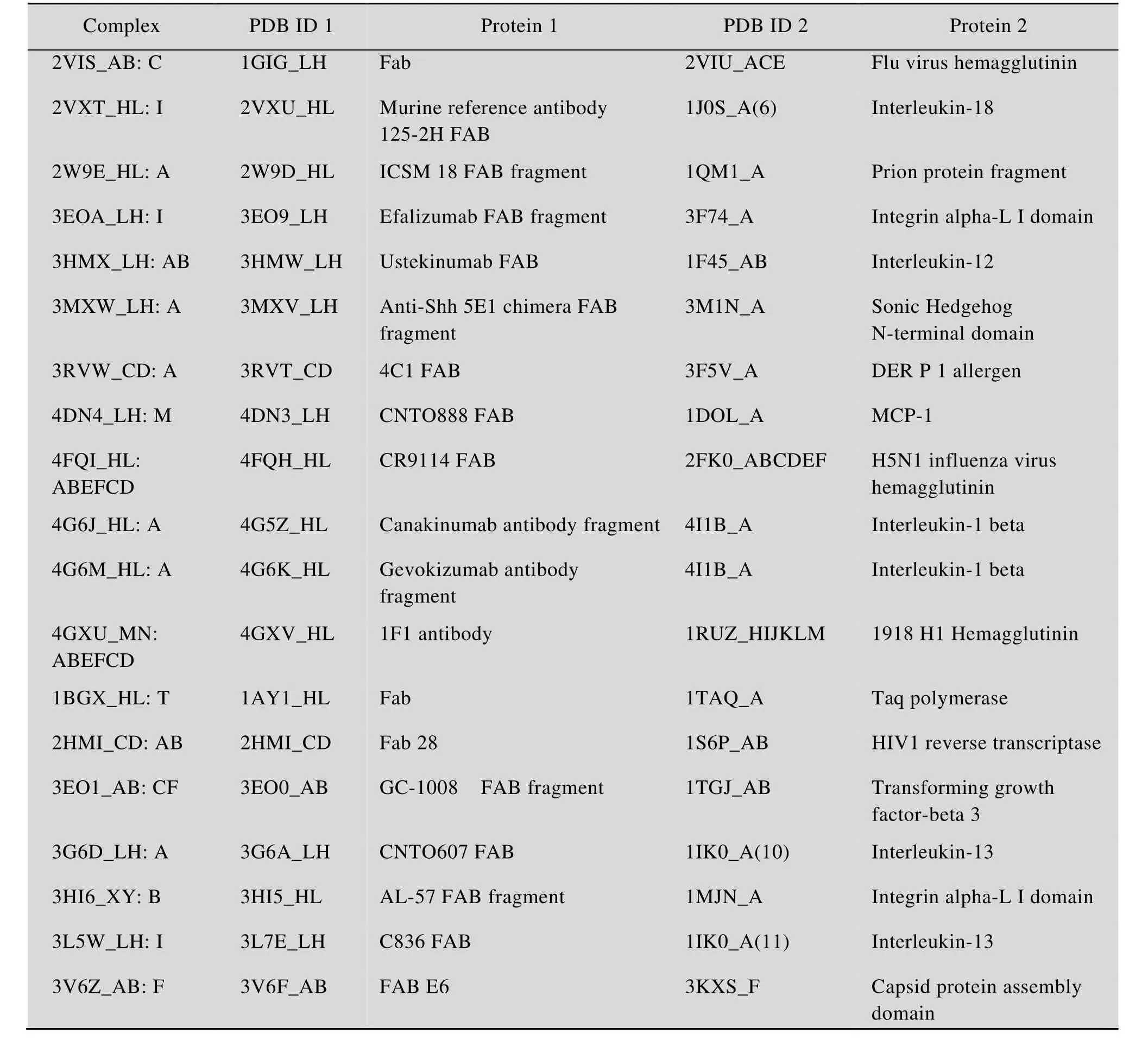
表1 训练集的19个抗原-抗体复合物Table 1 Training dataset of antigen-antibody complexes (total number of complexes=19)
1.5 二值资料多重logistic回归分析
准备好的训练集体系数据中含有一个定性变量:是否近天然结构 (即Y,取值为 0或 1),以及9个定量变量:①接触面面积;②接触面上氢键密度;③接触面上 cation-π密度;④EPII;⑤ZDock Score;⑥ZRank Score;⑦ZRank VdW;⑧ZRank Elec ; ⑨ZRank Solv 。 进行 证实 性 研究 ,以是否近天然结构 (Y) 为因变量,以上述9个定量变量为自变量,拟合logistic回归模型并采用逐步法筛选变量。P(Y=1)即P(This decoy is native-like)。自变量筛选以及 logistic回归分析使用专业的统计软件SAS 9.2完成。
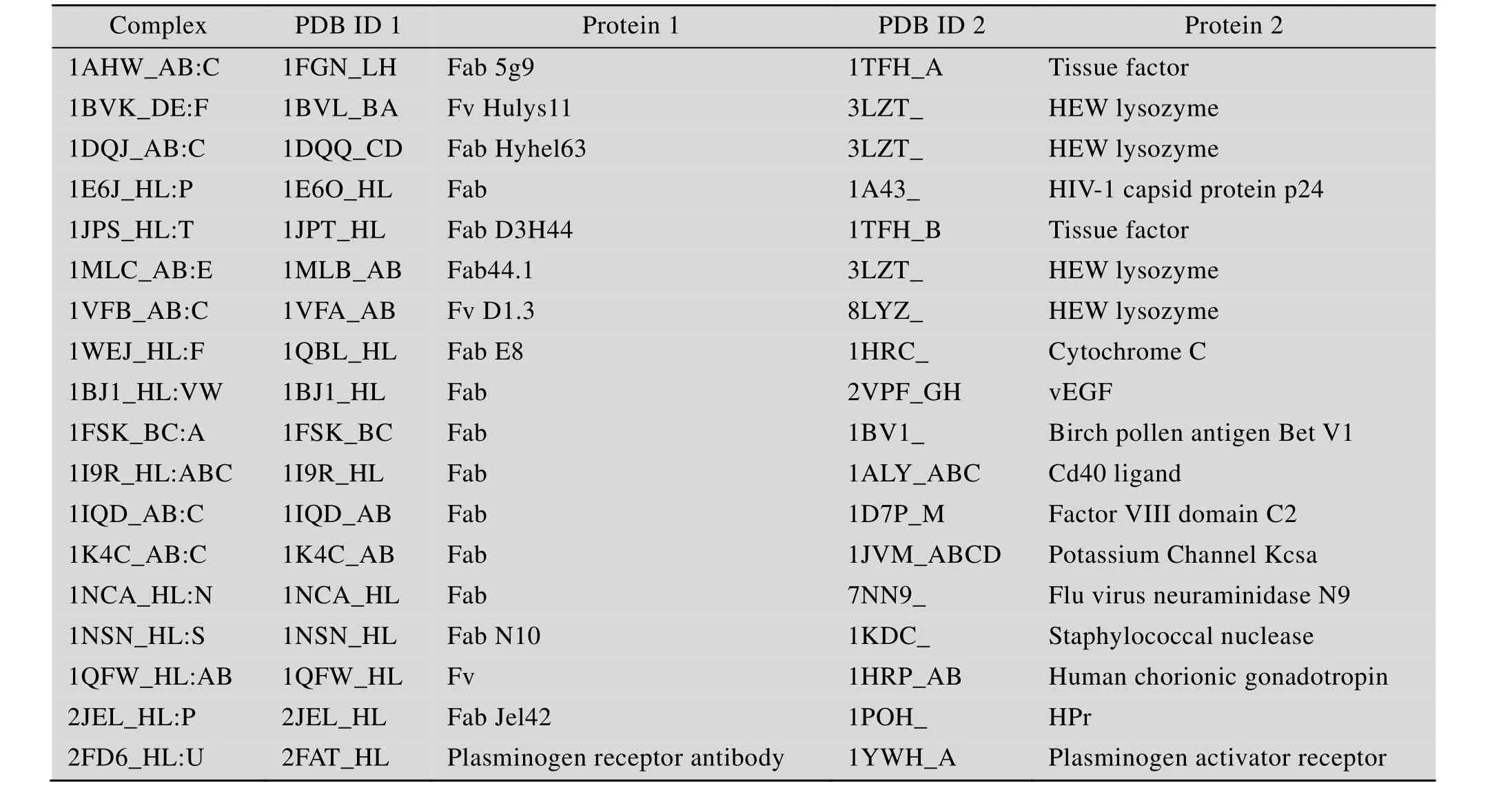
表2 测试集的18个抗原-抗体复合物Table 2 Testing dataset of antigen-antibody complexes (total number of complexes=18)
1.6 测试集对数学模型的验证
将测试集体系内各个对接模拟构象的接触面描述符和能量参数代入回归方程中,计算出各个构象模型的P(This decoy is native-like)值,并在各个体系内按P(This decoy is native-like)由高到低的降序对构象模型进行排序。统计各个测试集体系排名在前5位的对接构象模型中是否存在近天然结构,如果存在,则认为所建立的数学模型适用于该体系,成功筛选出该体系中的近天然结构。采用仅由 ZDock Score或ZRank Score打分排序的方法作为对照,采用相同的筛选成功与否的认定标准。该部分数据计算和统计等处理均由自行编写的Perl语言程序完成,BIOVIA Discovery Studio 4.5软件无相关功能。
1.7 预测埃博拉包膜蛋白与其中和抗体 4G7的结合模式
埃博拉病毒的包膜蛋白 (Glycoprotein,GP)在病毒入侵过程中扮演着关键的角色,是疫苗和抗体研究的重要靶标。抗体组合ZMapp (2G4,4G7,13C6)[16]是治疗埃博拉病毒感染的鸡尾酒疗法之一,组成ZMapp的中和抗体就结合于GP上的表位。使用从PDB数据库中下载的GP晶体结构[17](PDB ID: 3CSY) 和 4G7电镜结构[18](PDB ID: 5KEN) 在Discovery Studio 4.5软件中运用ZDOCK程序进行对接模拟。继而在生成的54 000个对接模拟构象中取出按ZDock Score排序在前5 000名的构象。计算出这5 000个构象的接触面描述符和能量参数并代入数学模型中得到P(This decoy is native-like),对P(This decoy is native-like)值最大的对接模拟构象使用基于机器学习的KFC2算法[19]预测抗原在接触面上的热点氨基酸 (关键氨基酸)。将筛选出的对接模拟结构与抗原-抗体复合物的电镜结构 (PDB ID: 5KEN) 相对比;将预测的抗原上的关键氨基酸与文献报道的实验数据相对比。
2 结果与分析
2.1 多重logistic回归分析得到数学模型
利用统计软件SAS 9.2进行回归分析,得到的有统计学意义的模型自变量:①接触面面积 (X1);②接触面上的氢键密度 (X2); ③ EPII (X3);④ZDock Score (X4),⑤ ZRank Score (X5),对各参数进行检验的P值均小于0.05。由此建立回归方程:
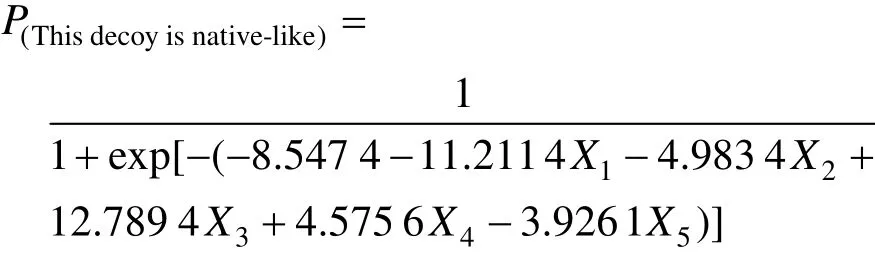
对整个模型进行假设检验,原假设是所有的回归系数都为 0,分别使用似然比、计分检验和Wald检验3种检验方法,3种方法的P值都小于0.05,可以认为该模型是成立的。ROC曲线的曲线下面积为0.994,预测概率和观察响应之间的关联性较强。
2.2 测试集对数学模型的验证效果
经测试,在全部18个测试体系中,模型成功筛选出了其中的15个体系中的近天然结构。作为对照,在全部18个测试体系中,ZDock Score打分方法成功筛选出了其中的6个体系中的近天然结构,ZRank Score打分方法仅成功筛选出了其中5个体系中的近天然结构 (表3)。对于18个测试体系 (共包含1 818个对接模拟构象),该数学模型筛选方法的有效性和成功率明显优于 ZDock Score打分方法和ZRank Score打分方法。
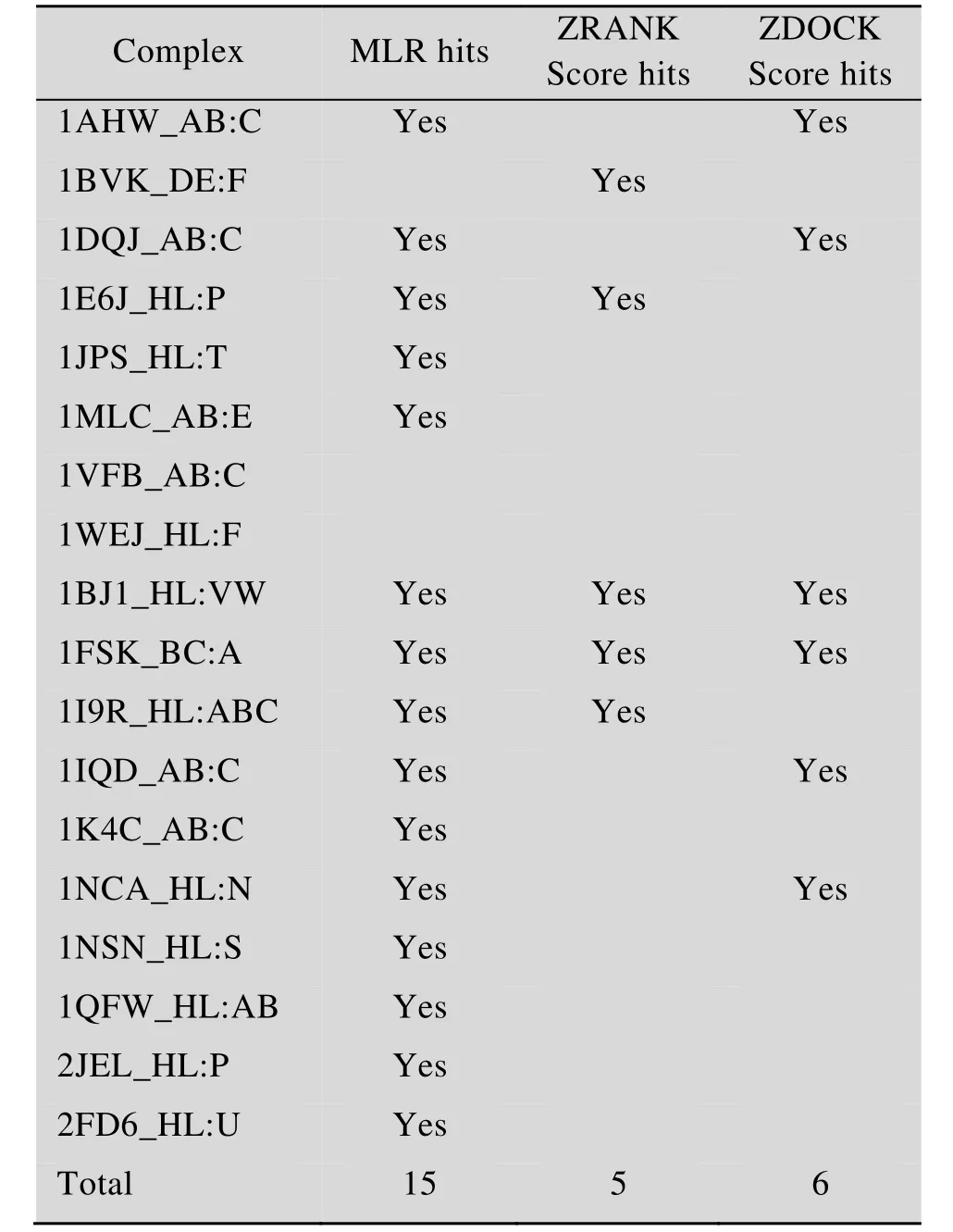
表3 测试集的预测结果Table 3 Prediction results of testing dataset (total number of complexes=19)
2.3 中和抗体4G7结合模式预测结果
文献[20]报道抗体4G7结合在GP Base上,抗原-抗体复合物电镜结构和实验数据表明Cys511、Asp552和Cys556是抗原上与抗体结合密切相关的关键氨基酸。应用数学模型得到的排序第一的对接构象模型,其抗原部分的 KFC2热点氨基酸 (关键氨基酸) 预测结果是 GP上的Asn506、Lys510、Cys511、Pro513、Asn550、Gln551、Asp552、Cys556,预测结果包含了全部3个文献报道的关键氨基酸 (粗体)。运用抗原-抗体分子对接并通过回归分析建立的数学模型筛选出与电镜结构相接近的近天然结构,有效预测出了抗体4G7与相应抗原的大致结合模式和以实验方法确定的关键氨基酸 (图1)。
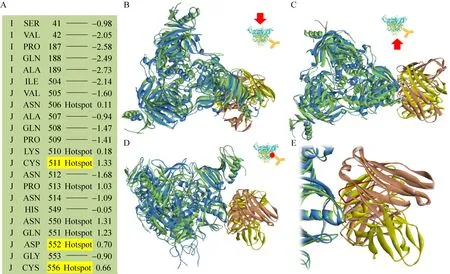
图1 埃博拉包膜蛋白中和抗体4G7结合表位的预测结果Fig. 1 Result of predicting epitope of anti-Ebola glycoprotein MAb 4G7. (A) Binding hot spots predicted by KFC Server. Residues critical for MAb 4G7 binding are highlighted in yellow. (B–E) Superposition of docking model and cryo-electron microscopy structure (PDB accession No. 5KEN) on fixed Ebola virus glycoprotein. Docking model is shown with glycoprotein colored green and MAb 4G7 colored yellow in which the heavy chain CDR3 loop is colored in purple. Cryo-electron microscopy structure is shown with glycoprotein colored blue and MAb 4G7 colored brown in which the heavy chain CDR3 loop is colored in red.
3 讨论
定量构效关系方法[21](Quantitative structure activity relationship,QSAR) 采用数理统计方法研究和揭示化合物活性与其分子结构或理化特性之间的定量变化规则,在小分子药物设计中有重要的应用。本文将QSAR的原理应用于抗原抗体对接模拟构象中近天然结构的筛选。即用数理统计方法抽提对接模拟构象的近天然程度与其抗原抗体接触面的理化特性、能量特性之间的定量变化规则。经回归分析,选定抗原-抗体接触面面积、接触面上氢键密度、EPII、ZDock Score和ZRank Score作为数学模型的自变量,建立多重 logistic回归方程。作为自变量的各参数由 BIOVIA Discovery Studio 4.5软件平台和自行编写的Perl语言程序计算得到,并需要进行标准化 (归一化)处理。使用所建立的多重 logistic回归方程指导从众多对接模拟构象中筛选出近天然结构取得较为理想的效果。在全部 18个测试集体系中,该方法可将其中 15个体系中的近天然结构排序在前5位,其中12个体系中的近天然结构排序在第1位,该方法对抗原-抗体近天然结构的筛选效果优于单纯使用ZRank Score或ZDock Score打分的排序筛选方法。基于该筛选方法在测试集验证中的良好表现,尝试将该方法应用于埃博拉病毒的包膜蛋白与其中和抗体4G7的结合模式预测。将 GP晶体结构与抗体 4G7的电镜结构进行ZDOCK对接,只有能够从生成的54 000个对接模拟构象中筛选出近天然结构,后续的热点氨基酸预测才能得到较为符合实际情况的结果。运用该数学模型计算后,取P(This decoy is native-like)值最大的对接模拟构象进行 KFC2热点氨基酸预测,预测出8个热点氨基酸,包括了文献报道的全部3个抗原上的关键氨基酸。热点氨基酸预测结果说明该对接模拟构象中抗原-抗体的结合方式 (尤其是接触面特征) 是接近真实情况或具有部分真实情况特点的。对抗体4G7大致结合模式的成功预测也在一定程度上说明了本文所提出的抗原-抗体近天然结构筛选方法具有可行性与实用性。
现有生命科学分子模拟软件平台,如Discovery Studio、HADDOCK[22]、RosettaDock[23]、AutoDock[24]、ClusPro[25]、PatchDock[26]、HDOCK[27]等,提供了分子对接程序及相应打分函数。可以通过蛋白质分子对接的方法研究蛋白质结合模式,但是对接过程中的全构象搜索产生成千上万的对接模拟构象,通用的打分函数很难满足我们进一步准确筛选出近天然结构的需要。不同类型蛋白质的结合具有各自特点和规律,可以通过对已有同类型蛋白质复合物共晶体结构的分析和统计得到相应的经验规律,而借鉴QSAR原理,运用多重回归分析的数学模型是将这些经验规律与已有打分函数相结合的有效途径。本文探索了蛋白质结合表面统计性、经验性特征与ZDOCK、ZRank打分函数的联合使用方法,证明了回归分析建立的数学模型用以打分排序和筛选出近天然对接模拟构象的可行性,为提高使用分子对接研究蛋白质结合模式的效率和准确性,提供了可行的思路和方法。
REFERENCES
[1]Huang SY, Bolser D, Liu HY, et al. Molecular modeling of the heterodimer of human CFTR’s nucleotide-binding domains using a protein-protein docking approach. J Mol Graph Modell, 2009, 27(7): 822–828.
[2]Loyau J, Didelot G, Malinge P, et al. Robust antibody-antigen complexes prediction generated by combining sequence analyses, mutagenesis,in vitroevolution, X-ray crystallography andin silicodocking. J Mol Biol, 2015, 427(16): 2647–2662.
[3]Li D, Xu J, Wang Z, et al. Epitope mapping reveals the binding mechanism of a functional antibody cross-reactive to both human and murine programmed death 1. MAbs, 2017, 9(4): 628–637.
[4]Lensink MF, Wodak SJ. Docking and scoring protein interactions: CAPRI 2009. Proteins, 2010, 78(15):3073–3084.
[5]Liang S, Meroueh SO, Wang G, et al. Consensus scoring for enriching near-native structures from protein-protein docking decoys. Proteins, 2009, 75(2): 397–403.
[6]Shimba N, Kamiya N, Nakamura H. Model building of antibody-antigen complex structures using GBSA scores.J Chem Inf Model, 2016, 56(10): 2005–2012.
[7]Tharakaraman K, Robinson LN, Hatas A, et al. Redesign of a cross-reactive antibody to dengue virus with broad-spectrum activity and increasedin vivopotency.Proc Natl Acad Sci USA, 2013, 110(17): E1555–E1564.
[8]Chen R, Li L, Weng ZP. ZDOCK: an initial-stage protein-docking algorithm. Proteins, 2003, 52(1): 80–87.
[9]Pierce B, Weng ZP. ZRANK: reranking protein docking predictions with an optimized energy function. Proteins,2007, 67(4): 1078–1086.
[10]Jones S, Thornton JM. Prediction of protein-protein interaction sites using patch analysis. J Mol Biol, 1997,272(1): 133–143.
[11]Crowley PB, Golovin A. Cation-π interactions in proteinprotein interfaces. Proteins, 2005, 59(2): 231–239.
[12]Accelrys Software Inc. Discovery Studio Modelling Environment. 2012.
[13]Méndez R, Leplae R, de Maria L, et al. Assessment of blind predictions of protein-protein interactions: current status of docking methods. Proteins, 2003, 52(1): 51–67.
[14]Hwang H, Vreven T, Janin J, et al. Protein-protein docking benchmark version 4.0. Proteins, 2010, 78(15):3111–3114.
[15]Rose PW, Prlić A, Altunkaya A, et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res, 2017, 45(D1):D271–D281.
[16]Qiu XG, Audet J, Lv M, et al. Two-mAb cocktail protects macaques against the Makona variant of Ebola virus. Sci Trans Med, 2016, 8(329): 329ra333.
[17]Lee JE, Fusco ML, Hessell AJ, et al. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature, 2008, 454(7201): 177–182.
[18]Pallesen J, Murin CD, de val N, et al. Structures of Ebola virus GP and sGP in complex with therapeutic antibodies. Nat Microbiol, 2016, 1(9): 16128.
[19]Zhu XL, Mitchell JC. KFC2: a knowledge-based hot spot prediction method based on interface solvation,atomic density, and plasticity features. Proteins, 2011,79(9): 2671–2683.
[20]Davidson E, Bryan C, Fong RH, et al. Mechanism of binding to Ebola virus glycoprotein by the ZMapp,ZMAb, and MB-003 cocktail antibodies. J Virol, 2015,89(21): 10982–10992.
[21]Cronin MTD, Basketter DA. Multivariate QSAR analysis of a skin sensitization database. SAR QSAR Environ Res, 1994, 2(3): 159–179.
[22]van Zundert GCP, Rodrigues J, Trellet M, et al. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J Mol Biol, 2016,428(4): 720–725.
[23]Chaudhury S, Berrondo M, Weitzner BD, et al.Benchmarking and analysis of protein docking performance in Rosetta v3.2. PLoS ONE, 2011, 6(8):e22477.
[24]Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem, 2009, 30(16):2785–2791.
[25]Kozakov D, Hall DR, Xia B, et al. The ClusPro web server for protein-protein docking. Nat Protoc, 2017,12(2): 255–278.
[26]Schneidman-Duhovny D, Inbar Y, Nussinov R, et al.PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res, 2005, 33(Web Server issue): W363–W367.
[27]Yan Y, Zhang D, Zhou P, et al. HDOCK: a web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res, 2017,45(W1): W365–W373.
