基于区域过滤的测序序列比对算法研究*
2018-05-23丁胜楠
丁胜楠,吴 鸣,徐 云
(1.中国科学技术大学 软件学院,安徽 合肥 230027;2.中国科学技术大学 计算机科学与技术学院,安徽 合肥 230027;3.安徽省高性能计算重点实验室,安徽 合肥 230027)
0 引言
DNA测序已经成为生物医学研究中不可或缺的工具,可直接应用于个性化医疗和基因变异导致的疾病研究。目前,二代测序技术,比较常见的有:罗氏454测序、Illumina等。而现在最常用的NGS技术就是Illumina测序技术,它可以确保在几十个小时内产生几百GB甚至上TB的测序数据,数据量特别巨大,完全能够满足高通量测序的通量要求。随着二代测序平台的不断发展,较第一代测序技术测序通量迅速增加,如目前常用的Illumina测序技术,它能在几个小时内产生数GB长度为100 bp(位点)的片段测序数据,将这些大量片段测序数据reads比对到参考基因组上,成为了测序序列比对算法面临的严峻挑战。
测序平台测出的短DNA序列片段(又称为reads)数量极高,达到数十亿个。在序列比对过程中,每个read在基因组中可能有一个或多个相似的片段,因此均需要在参考基因组中进行比对。同时,由于测序平台的局限性,每个测得的read可能会出现一些错误,以及基因组产生变异,如DNA序列中碱基可能会发生相应的插入、删除和替换(通常来说,read的这一类变化率小于等于5%)。因此,允许一定数量误配的字符串匹配是一个比较困难的问题,并会耗费大量的时间。以上问题导致基因序列比对在基因数据分析中占据了大量的时间。
测序序列比对分为找最好和找全比对两种应用问题。对于给定的一个编辑距离(插入、删除、替换)或海明距离的阈值k,找出每条测序序列(read,读数)在单个或多个基因组上满足阈值k的位置。如果需要找出所有位置,这类问题称为找全问题,对应的比对算法为找全算法(all-mapper);如果只需要找出分值(编辑距离或海明距离与位点质量的综合函数)最高或较高的一些位置,这类问题称为找最好问题,对应的比对算法为找最好算法(best-mapper)。找全测序序列比对是其中的重要问题,已有的mrFAST[1]、RazerS3[2]、GEM[3]、Bitmapper[4]、Bowtie2[5]、Masai[6]、Hobbes[7]等就是找全比对算法的代表。这些找全测序序列比对算法大多采用称为“种子-扩展(seed-and-extend)”的方法。该方法是先进行种子过滤,即将reads划分为若干个不重叠的长度为11 bp左右的种子,应用鸽巢原理确定出候选位置,对于人类基因组这样的候选位置往往会突现数千之多,严重增加了测序序列比对算法后续验证阶段的处理时间。因此,对于找全问题的测序序列比对算法,研究一种有效的过滤方法来减少候选位置来实现比对算法效率的提升,具有重要的研究意义和应用价值。
传统的种子过滤方法可以称为细粒度过滤法,这种方法产生的候选位置较多,有必要研究一种粗粒度过滤方法(比如区域过滤)来进一步地减少候选位置。文献[7]中提到的过滤算法,虽然上下边界限制条件比较好,但过滤是对每个候选位置的操作,而验证是对最终结果的操作,所以过滤需要尽可能快。这里采用了计数索引来加速过滤操作。Hobbes也采用了类似的方法,但其过滤未采用索引技术,增加了过滤的总时间。而本文的方法在较好的过滤效果和高效的计算中取得了一个平衡,综合来说,本文的区域过滤算法时间和性能较好。
1 相关工作
1.1 种子扩展方法(seed-and-extend)
种子扩展方法的第一步是将read分割为几个相对较短的,非重叠的seed。在查找过程中即使read中包含错误,但只要存在一个seed正确,则read的正确位置仍然可以被找到。而无错误的种子可以通过鸽巢原理进行选取,对于一个read,若允许最大误配数为k,那么由鸽巢原理得知,只需将其分为k+1个无重叠的seed,必然存在一个seed为正确的。对于一个不包含错误的seed来说,通过索引查找其在基因组中匹配的位置,也就得到了read的位置。
第二步,借助索引对seed进行快速地查找。在序列比对的过程中,seed的位置通常存储在对基因组预处理过的索引中(常用的索引有Hash索引和基于Burrows-Wheeler变换(BWT[8])的FM-index[9]),用来加快查找的速度。
最后一步,将查找到的位置返回基因组中做扩展验证。目前常用验证方法是动态规划算法(如Smith-Waterman[10]等)。通过扩展验证,检查read在基因组中匹配的质量,得出最终的位置,可以用于后续的生物学意义分析。
种子扩展方法流程如图1所示。
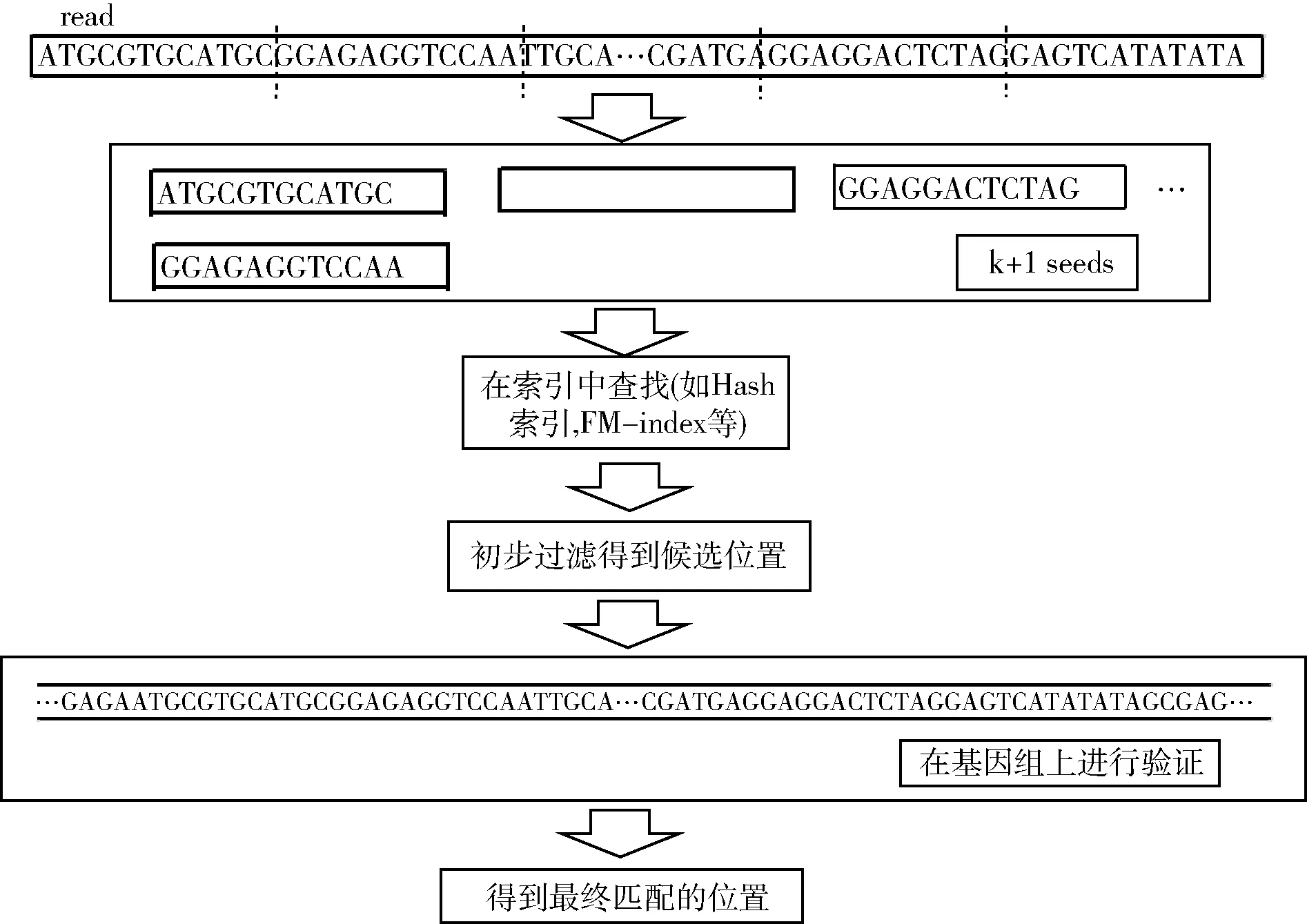
图1 种子扩展方法流程
从图1可以看出,将read切分成若干seed后,会产生很多重复以及错误的候选位置,而这些重复或者错误的候选位置也是需要过滤掉的种子。而参考基因组(待比对基因组)某一片段上碱基的个数是固定已知的,需要有一种方法将read中对应碱基含有误配的上下界找出来(且保证所找到的候选位置是无遗漏的),并以此做限定条件,去匹配含有编辑距离长度的基因片段里碱基的个数,进而筛选掉不在这个范围内的候选位置,来缩短比对和验证时间。
对于一个给定的read,假设允许的误配数为k,由鸽巢原理可知,将其分为k+1个seed,之后利用索引查找seed在基因组中的位置,鸽巢过滤后,再通过动态规划的验证扩展在基因组中进行精确比对,求出其最终匹配位置。
1.2 现有各种种子过滤方法
目前各种种子选取方法按种子长度可分为定长和变长,按种子数量可分为k+1、k+2等,其中k为编辑距离。不同的过滤方法往往带来非常不同的过滤效果,以下介绍近些年来种子过滤方法的研究和进展。
1.2.1定长种子过滤方法
FastHash提出,对于一个长度为100 bp的read,若seed的长度为12,则最多可选取8个种子。当k=5时,由鸽巢原理可知选取6个种子至少有1个种子一定是准确匹配的。先选取8个种子,先去除频次最大的2个种子,这样剩余的6个种子的候选位置就大大减少。由于种子的位置不同,可以组合出各种不同的k+1种子选取方案,Hobbes提出了动态规划选取k+1个定长种子,使其各种子的频次和为最小。Hobbes2在Hobbes的基础上,提出动态规划选取k+2频次最小的定长种子。由鸽巢原理易知,至少2个种子的位置是正确的,且这两个种子应该保持在reads上的准确相对位置,因此可对于k+2的种子进行相对位置再过滤,这样大大减少了候选位置。
1.2.2变长种子过滤方法
基于定长种子的过滤算法尽管时间复杂度低,但过滤后得到的位置数仍很多,而基于变长种子的过滤算法虽然时间消耗较定长过滤大,但仍在可接受范围内,而且可以找到最佳候选位置。基于变长种子的过滤算法如下:OSS提出了通过动态规划,选取k+1个频次最小的变长种子进行扩展验证;GEM提出了一种基于FM-index的变长种子的选取方法,先设定一个阈值t,从read的初始位置开始选取种子,若种子频率高于t,则扩展种子长度,否则,进行下一个种子的查找,直至选取k+1个种子。相比较变长种子过滤而言,由于定长种子速度快,种子位置多,对应的过滤效果更显著,因此本文基于定长种子过滤方法进行实验。
2 区域过滤方法设计
2.1 区域过滤方法的思想
为确保read在参考基因组的所有候选位置不漏选,制定了一个上下界来过滤,并对该上下界进行正确性证明,同时对该上下界进行优化选取。在此基础上设计出一种带有间隔且与read长度相当的滑动窗口,建立一种有效的滑动窗口方法。由于每个read包含的每种核苷酸(A/G/C/T)个数是固定的,经过k编辑距离后的read中每种核苷酸个数至多变化为k,最终目标是在这些滑动窗口中找出满足核苷酸个数变化的所有窗口。
2.2 方法描述
2.2.1构建滑动窗口核苷酸组成索引表
以线虫基因组为例,将其分成1 000份,每份有100 000长度的区段,设计方案中就以这100 000长度的区段为基因组作为示例,并将若干条长度为100 bp的read以编辑距离k=5比对到该区段上。滑动窗口设计如图2所示。为方便说明,以长度为110的滑动窗口为例。选取长度为110的滑动窗口,以间隔k=5进行滑动,这样在该区段上可以产生19 979个滑动窗口。
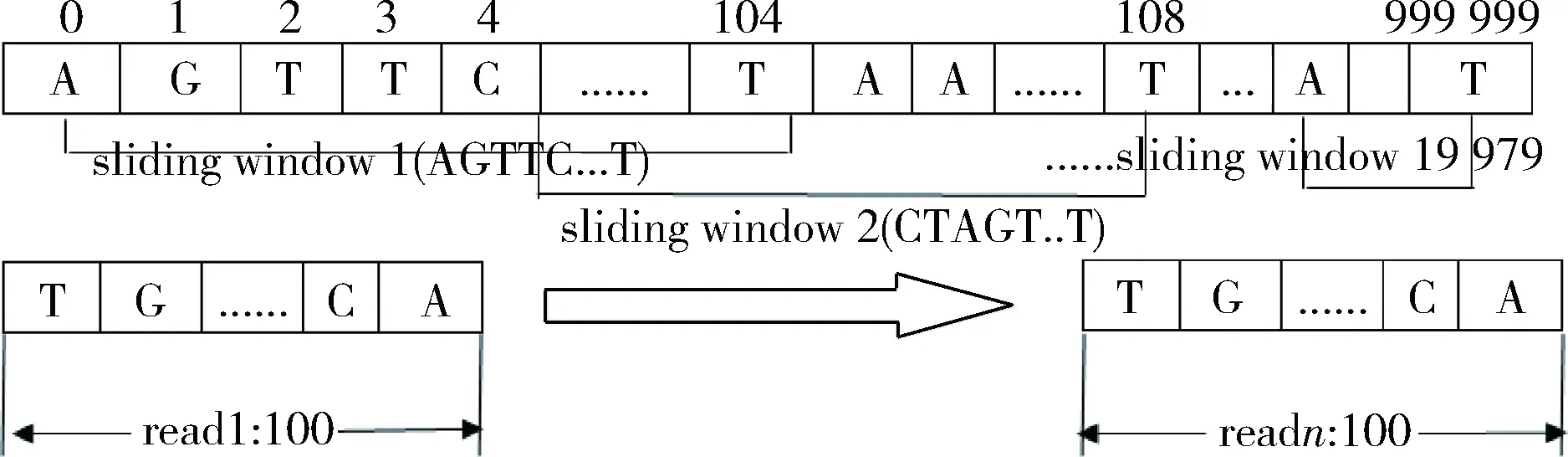
图2 滑动窗口示意图
按基因组建立如表1所示的A成分索引表,用于快速定位符合要求的滑动窗口。该索引表由每个滑动窗口的各个核苷酸成分个数组成。该索引表第1列为A成分个数,第2列为符合A成分个数的滑动窗口起始位置。

表1 核苷酸成分表
2.2.2索引表上检索符合筛选条件的滑动窗口
现假设ρr(x)为x在readr中出现的个数,x表示核苷酸A/C/G/T。那么在k编辑距离的情况下,可以得出如下过滤公式:
ρr(x)-3k≤ρw(x)≤ρr(x)+3k,x∈{A,C,G,T}
(1)
公式(1)是一个过滤公式组,每个核苷酸都应该将该公式作为一个相对独立的情形进行约束。
公式(1)是比较直观的,但过滤条件比较宽松。现在再考虑另一个相对紧凑些的过滤公式(2),过滤公式(2)是将4个核苷酸进行整体考虑。由于总碱基数目是固定的,每个碱基修改、删除、插入都有相对于另一个碱基的增减。比如:将A替换为C,这就意味着A减少1个而C增加1个。这样对于k次编辑距离,就得出过滤公式(2):
(2)
公式(2)的边界对公式(1)做了进一步的限定,在保证正确性的前提下对候选位置从整体进行了限定,使得过滤得到的区域更窄,进一步提升过滤率。
综上,正确候选位置需要满足上述条件,从而可以过滤错误的候选位置。
2.2.3与k+1过滤方法结合获取候选种子位置
将符合要求的区域(窗口)与seed位置进行重合检查,将在符合要求的区域(窗口)之外的seed位置过滤掉,从而减少种子候选集的大小。
2.2.4将区域过滤和BitMapper算法融合
将粗粒度和细粒度过滤后得到的种子候选位置应用到BitMapper算法中,去检测对应融合后的BitMapper算法时间性能的提升。具体做法为,对BitMapper比对算法中的过滤部分代码进行修改,在线虫基因组和人类基因组数据上进行计算实验,以检验算法的时间效率和精确度。
3 实验与分析
在线虫基因组和人类基因组上进行计算实验,检测区域过滤方法的过滤效果及其计算时间的提升。线虫基因组长度为1×108bp,相对人类基因组长度3×108bp而言规模要小些。两个不同规模的基因组能较为全面地反映区域过滤方法的效果。
3.1 过滤效果
过滤效果包括窗口和种子的过滤效果,通过过滤来减少在验证和比对过程所消耗的时间,作为性能是否提升的一个判别准则。在线虫基因组和人类基因组上的过滤效果分别如表2和表3所示,这里过滤后窗口(种子)比例对应的公式为:过滤后窗口(种子)数/过滤前窗口(种子)数。
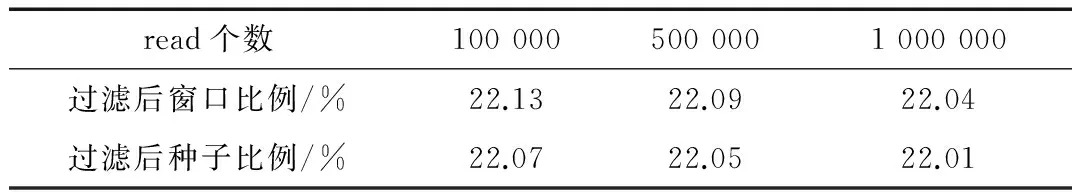
表2 线虫基因组过滤结果
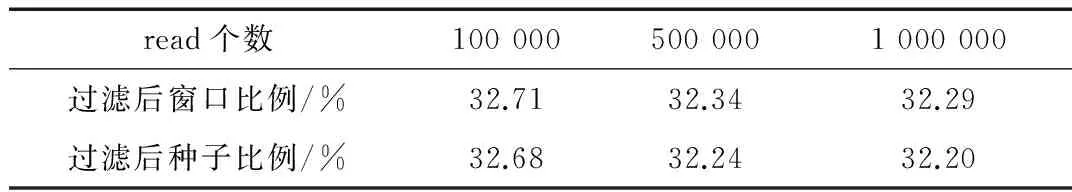
表3 人类基因组过滤结果
从表2可以看出,区域过滤方法在线虫基因组上是有效的,并且种子过滤效果要比窗口过滤的效果略好。从表3可以看出,区域过滤方法在人类基因组上也同样有效,相比于线虫基因组上效果要差些,主要原因是线虫基因组的结构性比人类基因组的结构性强些。
3.2 基于区域过滤的BitMapper算法时间性能
将区域过滤方法融合到原BitMapper算法中,对原BitMapper算法和融入区域过滤的BitMapper算法进行计算时间的比较,比较结果如表4和表5所示。
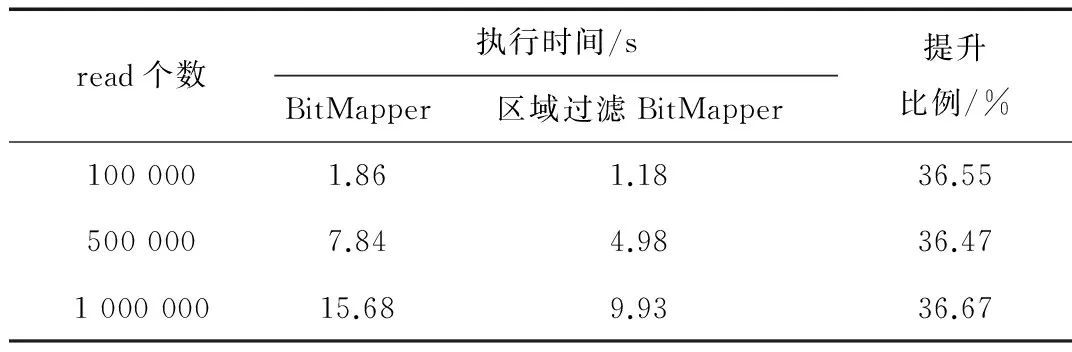
表4 线虫基因组上计算时间对比
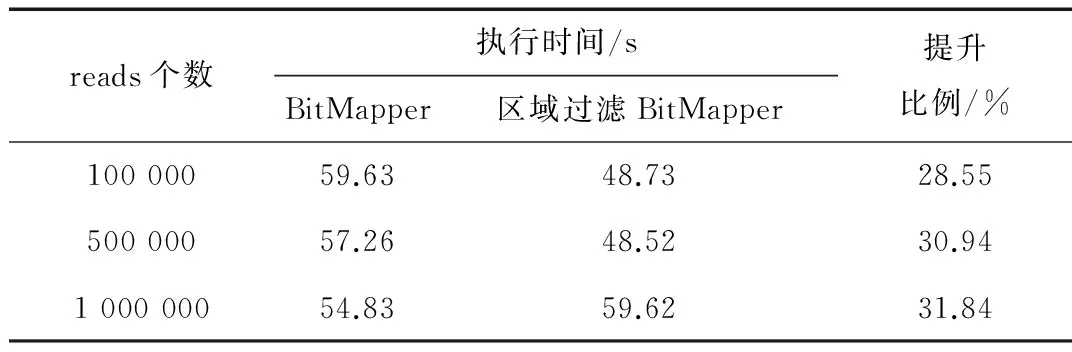
表5 人类基因组计算时间对比
尽管区域过滤方法比单一k+1过滤方法要消耗稍多些的过滤时间,但候选种子位置的显著减少,会大幅减少比对算法的验证时间,总体使得比对算法时间明显减少。表4表明线虫基因组计算时间提升了36%左右,表5表明人类基因组计算时间提升了30%左右。
4 结论
本文针对二代测序序列比对算法,提出了一种基于区域的粗粒度过滤方法。将该方法应用于线虫和人类基因组上,使得找全序列比对BitMapper算法有明显的时间性能提升。下一步的工作将考虑如何设计更精巧的索引,从而加快过滤速度,进一步提升比对算法的时间性能。
参考文献
[1] MARSCHALL T, MARZ M, ABEEL T, et al. Computational pan-genomics: status, promises and challenges[J]. bioRxiv, 2016: 043430.
[2] 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes[J]. Nature, 2012, 491(7422): 56-65.
[3] BALL M P, THAKURIA J V, ZARANEK A W, et al. A public resource facilitating clinical use of genomes[J]. Proceedings of the National Academy of Sciences, 2012, 109(30): 11920-11927.
[4] LEVY S, SUTTON G, NG P C, et al. The diploid genome sequence of an individual human[J]. PLoS Biol, 2007, 5(10): e254.
[5] DANECEK P, AUTON A, ABECASIS G, et al. The variant call format and VCFtools[J]. Bioinformatics, 2011, 27(15): 2156-2158.
[6] MARCO-SOLA S, SAMMETH M, GUIGR, et al. The GEM mapper: fast, accurate and versatile alignment by filtration[J]. Nature Methods, 2012, 9(12): 1185-1188.
[7] LI H, DURBIN R. Fast and accurate short read alignment with Burrows-Wheeler transform[J]. Bioinformatics, 2009, 25(14): 1754-1760.
[8] RAHN R, WEESE D, REINERT K. Journaled string tree-a scalable data structure for analyzing thousands of similar genomes on your laptop[J]. Bioinformatics, 2014, 30(24): 3499-3505.
[9] HUANG L, POPIC V, BATZOGLOU S. Short read alignment with populations of genomes[J]. Bioinformatics, 2013, 29(13): i361-i370.
[10] DANEK A, DEOROWICZ S, GRABOWSKI S. Indexes of large genome collections on a PC[J]. PloS One, 2014, 9(10): e109384.
