儿茶酚硼烷及其衍生物在有机反应中的应用进展
2018-05-04王合营
姜 涛,王合营, ,许 芳, ,崔 崑,马 志
(1. 天津科技大学化工与材料学院,天津 300457;2. 中国科学院上海有机化学研究所,上海 200032)
硼烷及其衍生物在硼氢化还原反应当中发挥着重要作用[1-4],相比于硼烷,硼烷衍生物在有机反应中的活性较低,但在反应的区域选择性和立体选择性方面有着独特的优势,因而对于硼烷衍生物的合成及其在有机反应中的应用一直受到相关研究人员的关注.儿茶酚硼烷(catecholborane,HBcat)是一种单氢的硼烷衍生物,最早是由 Newsom 等[5]在 1968年报道的.儿茶酚硼烷的合成方法简单,它与硼烷在反应活性和稳定性上具有较大的区别,儿茶酚硼烷在参与有机反应时的区域选择性和立体选择性较高.与其他硼烷衍生物相比,儿茶酚硼烷还具有如下一些优点:(1)儿茶酚硼烷在室温下是液体,也可以在无溶剂的条件下直接使用;(2)儿茶酚硼烷可以溶解并稳定于大多数非质子溶剂中(例如:苯、甲苯、氯仿、乙醚和己烷等);(3)儿茶酚硼烷在干燥的空气中性质稳定,仅在潮湿的空气中会发生水解反应生成没有氢化活性的化合物;(4)儿茶酚硼烷具有长期的贮存稳定性,可以在 0,℃的条件下保存 1年以上而不发生变化.本文对近年来儿茶酚硼烷及其衍生物在有机反应中的应用进展进行评述.
1 儿茶酚硼烷的合成方法
儿茶酚硼烷的制备方法有很多种,最常用的有以下几类(图1[5-10]).
Newsom 等[5]在 1968年首次报道了儿茶酚硼烷的合成,是由三丁基氢化锡与氯化儿茶酚硼烷在90,℃的条件下反应得到的(图 1(a)).该合成路线的反应速率适中,但反应产率较低,仅为42%,.
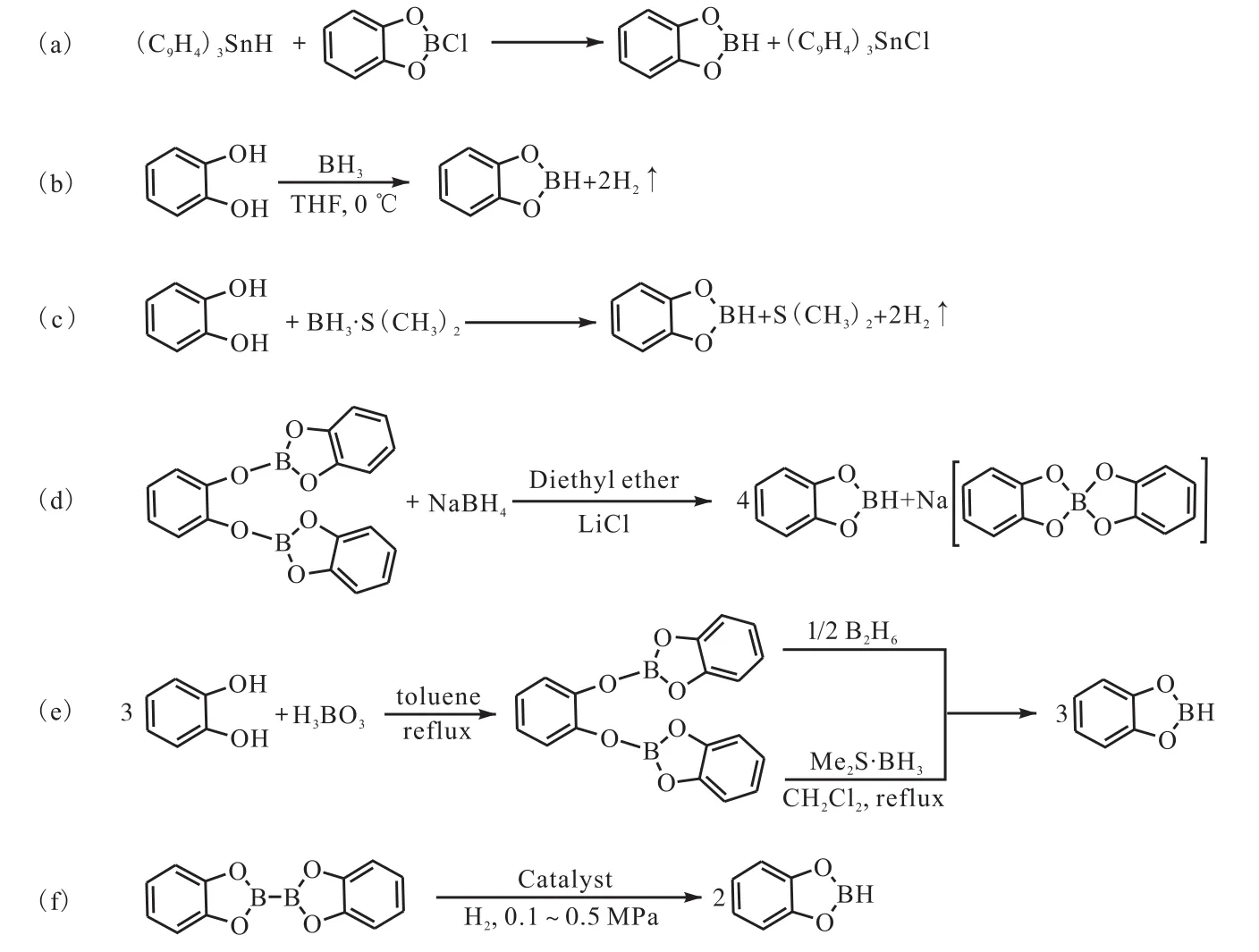
图1 儿茶酚硼烷的合成Fig. 1 Syntheis of catecholborane
1971年,Brown等[6]以四氢呋喃(THF)为溶剂,使用儿茶酚与硼烷制备儿茶酚硼烷(图 1(b)).此反应需在 0,℃的条件下将反应物混合,然后升温至25,℃,30,min内反应产率可以达到 80%,.虽然此合成路线在反应转化率和反应速率方面具有很大的优势,但是其反应产物中的THF很难完全除去.
鉴于儿茶酚硼烷产物中THF难以完全除去的缺点,Brown 等[7]采用硼烷-二甲基硫醚(BH3⋅S(CH3)2)和儿茶酚作为原料,并改用乙醚作为反应溶剂制备儿茶酚硼烷(图 1(c)).此方法解决了儿茶酚硼烷中溶剂难以除净的问题,但是该合成路线的反应速率和反应转化率都有所下降.
Männig等[8]选择邻三亚苯基双硼酸盐和 NaBH4作为反应原料,以乙醚为溶剂,LiCl为添加剂,利用球磨法合成得到儿茶酚硼烷(图 1(d)).其反应转化率为 80%,,分离产率可以达到 75%,.该合成路线在保持较高反应转化率的同时,也解决了儿茶酚硼烷产物中溶剂残留的问题.此后,Kanth等[9]报道了一系列经济、高效、环保的儿茶酚硼烷合成方法,即利用邻三亚苯基双硼酸盐与 B2H6和 BH3⋅S(CH3)2分别在不同溶剂中进行反应(图 1(e)).儿茶酚硼烷的分离产率提升到85%,,纯度则高达97%,.
2013年,Braunschweig等[10]将二硼酸盐化合物在非均相金属催化剂的作用下进行 B—B键的高压氢裂解反应,合成了包含儿茶酚硼烷(图 1(f))在内的三类单氢硼烷.其中以钯类催化剂的催化效果最为理想,此类催化剂可以在常温条件下高效地催化二硼酸盐当中的B—B键断裂,随后与氢气进行反应生成儿茶酚硼烷.
2 儿茶酚硼烷参与的烯烃硼氢化反应
烯烃的硼氢化反应是有机合成中的重要化学反应,因为硼氢化反应生成的产物可以非常方便灵活地再次被转化成为含有不同官能团的产物.硼氢化反应被定义为硼烷或者至少含有一个 B—H键的硼烷衍生物在碳碳双键上加成,生成有机硼化合物的反应.在经典的硼氢化反应过程中,B—H键中的 B原子总是加成在不饱和键上取代基较少的一边,形式上生成以反马氏加成为主的有机硼化合物.作为一种单氢硼烷,儿茶酚硼烷在 25,℃的条件下与烯烃的反应非常缓慢,但是在儿茶酚硼烷过量 10%,、100,℃下则会快速高效地与烯烃进行硼氢化反应,生成相应的烷基儿茶酚硼烷,参与反应的烯烃的转化率超过90%,.过渡金属催化剂的使用使得儿茶酚硼烷参与的硼氢化反应可以在室温下进行,同时催化剂也可以改变儿茶酚硼烷在碳碳不饱和键上的插入点[5-6].
催化剂的使用使得儿茶酚硼烷在低温条件下反应活性低、区域选择性和立体选择性差的缺点得到改善.Männig等[11]率先报道了多面体硼烷在惰性金属催化剂的作用下与烯烃和炔烃的反应,儿茶酚硼烷可以在催化剂的作用下与烯烃进行在无催化剂条件下所不能完成的化学选择性硼氢化反应(图2[11]).在铑类催化剂的作用下,烯酮类化合物当中的碳碳双键与儿茶酚硼烷进行硼氢化反应,随后使用过氧化氢处理目标产物,转化率可达65%,.
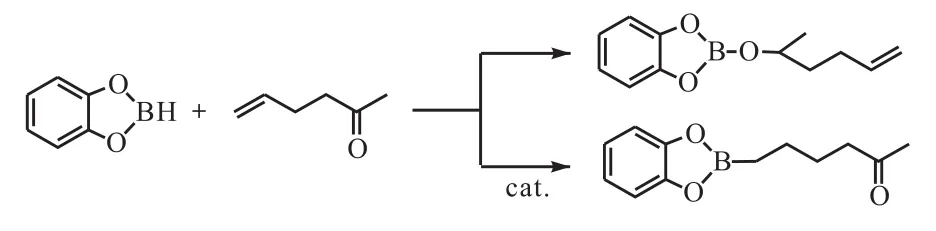
图2 儿茶酚硼烷对5-己烯-2-酮的选择性硼氢化Fig. 2 Selective hydroboration of 5-hexen-2-one using HBcat
Zaidlewicz等[12]报道了共轭二烯烃和炔烃与儿茶酚硼烷的硼氢化反应.共轭二烯烃在钴类催化剂的作用下可与儿茶酚硼烷进行 1,2-加成和 1,4-加成反应,但是在镍类催化剂的作用下几乎只进行 1,2-加成反应(图3[12]).
Vogels等[13-14]鉴于儿茶酚硼烷的性质对惰性金属催化硼氢化反应的机理进行了探索(图 4[14]),结果显示没有位阻的苯乙烯衍生物与儿茶酚硼烷在催化剂的作用下生成特定的芳基儿茶酚硼烷,但是在与具有不同取代基的苯乙烯反应时则生成没有分布规律的儿茶酚硼烷衍生物,Lillo等[15]对儿茶酚硼烷与不同烯烃反应产物的硼氢化点进行了细致的研究.
儿茶酚硼烷的硼氢化反应温度较高,而将过渡金属催化剂用于硼氢化反应可以降低反应温度,并且使得儿茶酚硼烷在反应过程中具有区域选择性、立体选择性和化学选择性来弥补无催化剂条件下硼氢化反应的不足是当前研究的热点.Vogels等[16]对儿茶酚硼烷与烯丙胺以及烯丙基胺衍生物在有催化剂和无催化剂的条件下的原位反应进行了研究,探索了在没有保护基团的条件下儿茶酚硼烷与胺基化合物反应产物的组成(图5[16]).
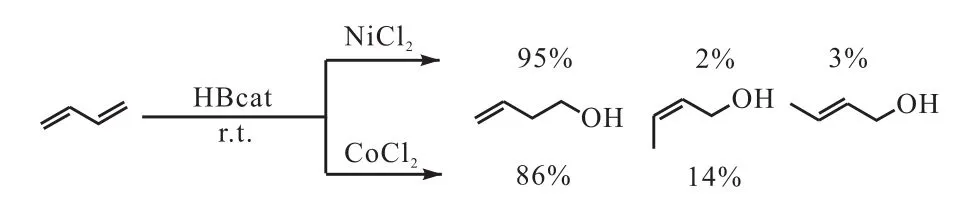
图3 共轭二烯烃的催化硼氢化Fig. 3 Catalytic hydroboration of conjugated diene using HBcat
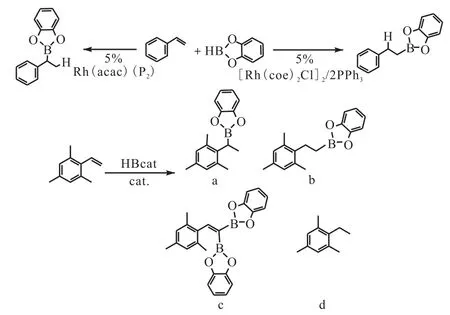
图4 苯乙烯催化硼氢化Fig. 4 Catalytic hydroboration of styrene using HBcat
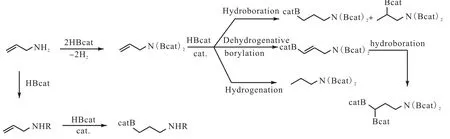
图5 烯丙基胺/亚胺的催化硼氢化Fig. 5 Catalytic hydroborations of allylamine and allylimines with HBcat
Brown等[17]研究了包括儿茶酚硼烷在内的不同硼氢化试剂与直链烯烃之间的反应,结果发现直链末端烯烃与硼氢化试剂进行反应的速率与直链长度无关,只与直链烷烃 α位上的取代基有关.儿茶酚硼烷在 THF中与丁烯进行硼氢化反应,94%,硼原子在反应过程中连接到末端碳原子上;在与异丁基乙烯反应的过程当中,99%,的硼原子连接到末端碳原子上.然而,在儿茶酚硼烷与苯乙烯进行硼氢化反应的过程当中,只有 81%,的硼原子连接到末端碳原子上,这与苯乙烯当中苯环上的电子对儿茶酚硼烷的影响较小,只有少量的硼原子插入到苄基位置上的推测相违背,同时他们还使用降冰片烯与儿茶酚硼烷进行反应并对其立体定向选择性硼氢化进行了研究.
Goddard等[18]使用过量的儿茶酚硼烷在 100,℃的条件下与烯烃进行反应制备烷基儿茶酚硼烷,随后将烷基儿茶酚硼烷与叠氮甲烷进行同系化反应生成三甲基硅烷基中间体,再用碱性过氧化氢与氟化正丁基胺处理生成烷基醇,在此过程当中没有氢氧化锂参与反应,同时还避免了一氧化碳的使用(图6[18]).
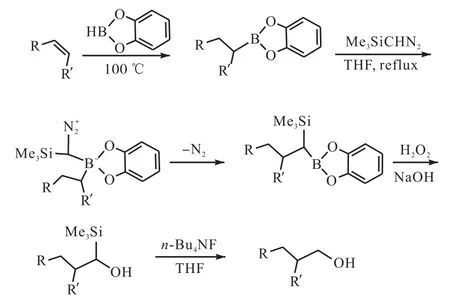
图6 烯烃的高温硼氢化及其在烷基醇制备中的应用Fig. 6 Hydroboration of alkene at elevated temperature with HBcat and its application in the syntheses of alcohols
3 烷基儿茶酚硼烷的应用
3.1 烷基儿茶酚硼烷
烷基儿茶酚硼烷是一种非常好的链转移试剂和自由基前驱体,它可以在自由基链增长反应过程当中释放出伯、仲、叔烷基自由基与以氧原子或其他原子为中心的自由基进行反应,从而使得共轭加成反应、烯丙基化反应、烯基化反应和炔基化反应能够顺利进行.这也使得科研人员对烷基儿茶酚硼烷在各种反应当中的应用较为感兴趣.有机硼烷在与以氧原子为自由基中心的自由基进行反应的过程当中,硼原子上烷基取代基的反应活性由有机硼烷分子的路易斯酸性所决定.Baban等[19]报道了不同硼烷的反应活性:R3B>R2BOR>RB(OR)2(R 为烷基).由于硼原子和氧原子π键的作用,硼酸酯当中烷基与烷氧基自由基反应的活性要低于三烷基硼当中烷基,但烷基儿茶酚硼烷当中烷基的活性却与人们的预期有所不同.它对氧气非常敏感并且能够快速地与烷氧基自由基进行反应,甲基儿茶酚硼烷可以与烷氧基自由基进行反应,并通过苯环上的电子离域作用使得烷氧自由基稳定地连接到硼原子环上面(图7[19]).
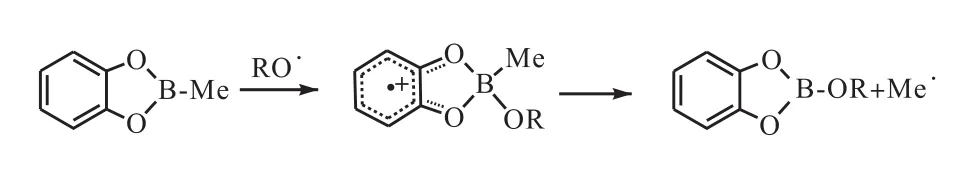
图7 烷基儿茶酚硼烷自由基化反应Fig. 7 Radical release of alkylcatecholborane
烯烃与儿茶酚硼烷通过硼氢化反应制备的烷基儿茶酚硼烷当中的烷基链比三烷基硼烷当中的烷基链的反应活性要高,烷基儿茶酚硼烷可以通过选择不同的烯烃使得烷基儿茶酚硼烷在自由基反应过程当中释放出相应的烷基自由基.烷基儿茶酚硼烷在与以杂原子为中心的自由基的反应过程当中释放出烷基自由基,反应过程当中硼氧键断裂使得杂原子为中心的自由基连接到硼原子上,但是接着会进行不可逆的反应,释放出烷基自由基(图8[19]).
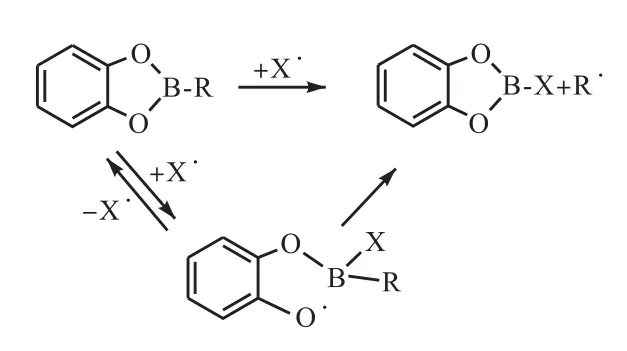
图8 烷基儿茶酚硼烷释放烷基自由基机理Fig. 8 Mechanism of alkyl radical releasing from alkylcatecholborane
3.2 烷基儿茶酚硼烷的TEMPO氧化
Studer 课题组[20-22]采用 2,2,6,6-四甲基哌啶-N-氧自由基(TEMPO)作为氧化剂和封端基团,将烯醇类儿茶酚硼烷化合物氧化生成含有2,2,6,6-四甲基哌啶-N-氧基团的酮类化合物(图 9[21]).烷基儿茶酚硼烷通过与 2,2,6,6-四甲基哌啶-N-氧自由基作用生成以氧原子为中心的自由基,此类自由基可以与烯酮类化合物进行可控氮氧自由基聚合反应.
Ollivier等[23]报道了烯烃与儿茶酚硼烷进行硼氢化反应生成对应的烷基儿茶酚硼烷,随后被处理生成相应烷氧基胺.在反应过程当中,N,N-二甲基已酰胺作为烯烃硼氢化过程中的催化剂,等物质的量的TEMPO与烷基儿茶酚硼烷反应释放出等物质的量的烷基自由基,另外等物质的量的TEMPO与烷基自由基相互作用生成烷氧基胺.反应产物的立体化学结构与所期望的自由基反应相吻合,两个自由基选择性地进行反应过程当中,TEMPO巨大的位阻是控制此立体选择性反应的关键(图10[23]).
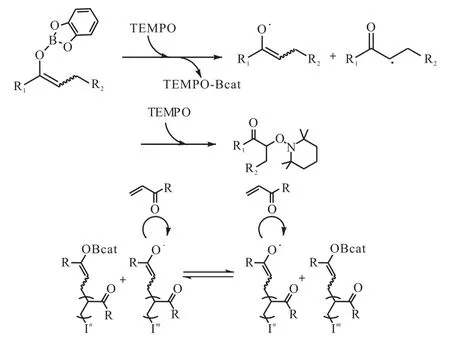
图9 烷基儿茶酚硼烷烷氧胺自由基氧化Fig. 9 Catecholboranes as precursors for alkoxyamines and nitroxide-mediated polymerization

图10 烷基儿茶酚硼烷自由基化Fig. 10 Radical release of alkylcatecholborane
对于较为特别的反应基质,使用自由基为中间介质所得的有机硼烷氧化产物与使用普通氧化方法生成的产物不同,例如,2-蒈烯进行硼氢化/氧化反应生成二环醇.被TEMPO氧化过的烷基儿茶酚硼烷经由环丙烷基自由基开环反应,再用锌可将其还原生成单环叔醇.然而,TEMPO与自由基的初始反应速率较快,所以此类反应只适用于反应活性较高的烯烃.
值得注意的是,只有少数的自由基羟基化反应被报道出来,一方面是由于反应选择性较低,另一方面其反应产物产率较低.Cadot等[24]借助于儿茶酚硼烷的硼氢化特性,通过烯烃的硼氢化反应生成的烷基儿茶酚烷,然后使用氧气或是 TEMPO进行处理,随后通过还原反应将其转化为相应的醇.此反应的反应条件较为温和,操作也较为简便,并且该反应还可以延伸到环化反应和羟基化过程的研究中.
通过硼氢化反应制备的烷基儿茶酚硼烷可以在TEMPO的作用下生成含有不稳定CON键的烷氧基胺.这种能够作为 NMP引发剂的烷氧基胺首次被Wagner等[25]报道,他们将烷基儿茶酚硼烷作为制备含有烷氧基胺的前驱体,随后将烷氧基胺作为大分子引发剂/调节剂,选用苯乙烯和丙烯酸丁酯作为单体通过氮氧自由基反应制备梳形和星形聚合物.
3.3 烯酮和二烯醛共轭加成反应
自由基反应是有机合成当中的重要反应,通常被应用于合成天然产物,却很少被应用于工业生产,这主要是因为在自由基反应过程当中要引入有毒且难以去除的有机锡试剂.近 10年来,越来越多的科学家投身研究不需要有机锡参与的自由基反应.有机硼烷可以在自由基反应过程当中生成伯、仲、叔烷基自由基和无毒的硼酸衍生物,因而成为可供选择的替代有机锡的自由基前驱体.
Renaud课题组[26-29]将烷基儿茶酚硼烷作为烷基自由基前驱体,并将其应用于与一系列的烯酮进行加成反应.在随后的研究当中,他们将其作为反应物与不饱和酯类化合物进行分子内环化反应,同时烷基儿茶酚硼烷也可以在一定的条件下释放出烷基自由基与2-苯磺酰类化合物进行反应(图11[27-29]).
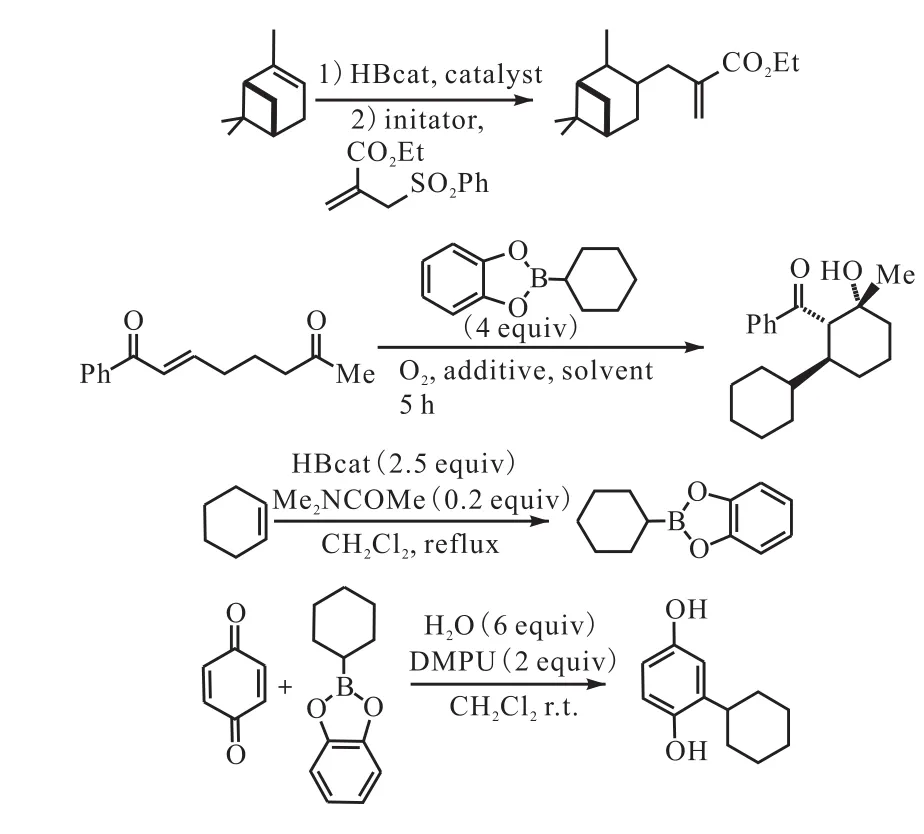
图11 烷基儿茶酚硼烷共轭加成到烯酮Fig. 11 Radical addition of modified alkylcatecholboranes onto enones
依据 Brown等[30]先前的报道,三烷基硼烷在氧气的作用下释放出烷基自由基,与烯酮和烯醛进行反应,然后进行水解生成硼烯醇酯,但这种产生自由基的方法并未广泛应用于合成,且此方法有一个严重缺点:三烷基硼烷当中只有一个烷基能够转化为烷基自由基.Ollivier等[31]通过实验证明,烷基儿茶酚硼烷与烯烃进行硼氢化反应生成的烷基儿茶酚硼烷可以在氧气存在的条件下释放出烷基自由基,与烯酮或者烯醛进行共轭加成反应(图12[31]).
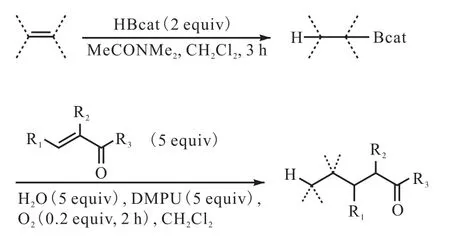
图12 烷基儿茶酚硼烷共轭加成Fig. 12 Conjugate addition of alkylcatecholborane to enones
自由基反应在分子内和分子间碳碳键的形成反应当中扮演着重要的角色,锡氢化物加成到活性烯烃是自由基加成反应当中常用的反应之一,然而当此反应应用于药物合成时,产物中的有机锡残留物难以全部去除,Ollivier等[32]使用甲氧基吡啶-2-硫酮-N-氧酸甲酯(PTOC-OMe)作为链转移试剂,在 150,W 钨灯的照射下与烷基儿茶酚硼烷进行反应生成相应的含硫衍生物和甲氧羰氧基自由基(图 13[32]),产物当中的含硫官能团可以被除去或是被转化为其他官能团.由于此反应需要先将反应原料进行硼氢化,因此该反应可以利用儿茶酚硼烷的化学选择性和区域选择性制备光学活性材料和对映异构体化合物.
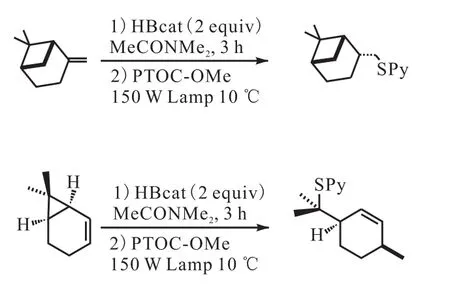
图13 自由基磺化Fig. 13 Radical sulfurization with PTOC-OMe
此反应不仅适用于 2-蒈烯,而且儿茶酚硼烷与活性烯烃的硼氢化共轭加成产物同样适用于以下体系(图 14[32]).PTOC-OMe在光照的条件下生成甲氧羰基自由基和甲氧基自由基来引发反应,生成的自由基与烷基儿茶酚硼烷反应得到亲核烷基,自由基加合物当中的取代基具有一些亲电特性,因此可以快速地与 PTOC-OMe当中的富电子硫代羰基进行反应,因此该反应可以作为锡氢化物氢化反应的替代反应.
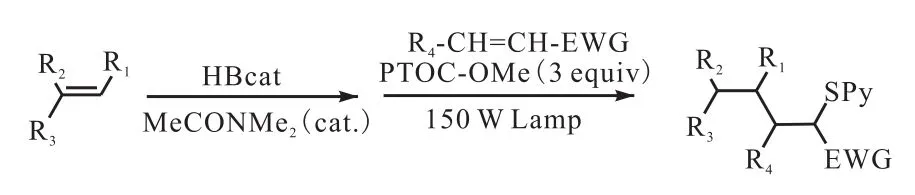
图14 烷基儿茶酚硼烷共轭加成到烯烃Fig. 14 PTOC-OMe-mediated conjugate addition of alkylcatecholborane to activated alkenes
3.4 自由基烯丙基化
烷基儿茶酚硼烷在过氧化氢或者其他氧化试剂的作用下生成醇类化合物,Renaud课题组[33-35]发现烷基儿茶酚硼烷在氧气和过氧化氢的作用下释放出烷基自由基、烷氧基自由基和磺酰基自由基,可在共轭加成反应和烯丙基化作用过程当中作为中间体使得碳碳键连接.针对此类反应,他们研究了不同的苯磺酰基反应物对反应结果的影响,此后针对 α-蒎烯类化合物与儿茶酚硼烷的硼氢化产物又进行了深入地探索.
烷基儿茶酚硼烷在氧气和过氧化物的引发下生成烷基自由基,Pozzi等[33]选用甲醇作为还原性试剂,甲醇分子当中羟基基团上的氢原子连接到烷基自由基上得到烯烃还原产物.随后他们又研究了不同烷基儿茶酚硼烷与烯丙基苯磺酰类化合物进行自由基反应,烷基儿茶酚硼烷在氧气的作用下生成烷基自由基与烯烃进行反应生成新的烷基自由基,新形成的烷基自由基与烯丙基苯磺酰类化合物反应生成新的烯丙基类化合物.
Schaffer等[36]借鉴 Davies等[37]使用苯磺酰基溴与三丁基硼烷反应制备丁基溴的反应原理,将其应用于烯丙基亚砜与烷基儿茶酚硼烷进行反应制备烯丙基化产物(图 15[36]),二叔丁基连二次硝酸盐在二氯甲烷当中加热生成叔丁氧基自由基与烷基儿茶酚硼烷进行反应生成烷基自由基,随后烷基自由基与苯磺酰类化合物进行反应释放出苯磺酰自由基以维持此链式反应(图16[36]).
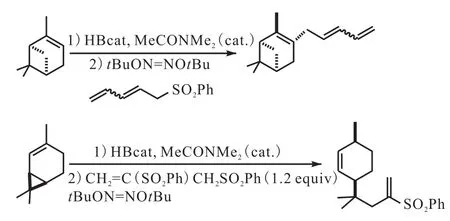
图15 烯丙基化反应引入二烯基Fig. 15 Introduction of a dienyl moiety using penta-2,4-dienyl phenyl sulfone

图16 自由基烯丙基化反应及其机理Fig. 16 Radical hydroallylation of alkenes and the mechanism
Garrett等[38]利用二叔丁基连二次硝酸盐的性质开发出了反应效率高、反应条件温和、经济性好的硼氢化/自由基反应,整个反应过程为一个完整的还原/烯丙基化反应.无取代基的烯丙基亚砜在相同的反应条件下同样能够进行反应,但生成的烯丙基化产物不稳定,产物产率在68%,~95%,.
4 儿茶酚硼烷参与的炔烃硼氢化反应
儿茶酚硼烷可以在温和的条件下与炔烃进行硼氢化反应生成重要的有机合成中间体.1-己炔和 3-己炔与儿茶酚硼烷在 68,℃的条件下分别反应 4,h和8,h后反应产率达到 90%,,但是在 70,℃的条件下与末端烯烃和分子内炔烃反应分别需要 1,h、2,h,相比之下,儿茶酚硼烷与末端炔烃反应的速率比与分子内炔烃反应的速率要快[39].无催化剂参与的儿茶酚硼烷硼氢化反应,反应速率较为缓慢,当反应体系当中引入溶剂时会进一步减缓反应速率,因此科学家期望通过引入催化剂使得硼氢化反应速率变得快速高效且反应条件更加温和.
Ohmura等[40]报道了末端炔烃在铑和铱类催化剂的作用下与儿茶酚硼烷进行硼氢化反应生成顺式烯烃基硼烷,在室温条件下 1-辛炔的硼氢化产物的收率为 34%,~94%,,随后又对不同的末端炔烃的硼氢化产物进行了研究.
Arase等[41]以二硼烷(二环己基硼烷、9-硼杂双环壬烷)为催化剂使儿茶酚硼烷与炔属烃在温和的条件下进行硼氢化反应,1-己炔与儿茶酚硼烷在室温条件下反应 2,h,1-己炔转化率为 75%,.在同样的反应条件下,儿茶酚硼烷与 1-己炔在二烷基硼烷的催化作用下进行硼氢化反应,反应转化率高达96%,.烯烃基儿茶酚硼烷衍生物是多种合成反应的重要中间体,Arase等通过水解、转换反应将烯烃基儿茶酚硼烷转化为对应的化合物(图17[41]).
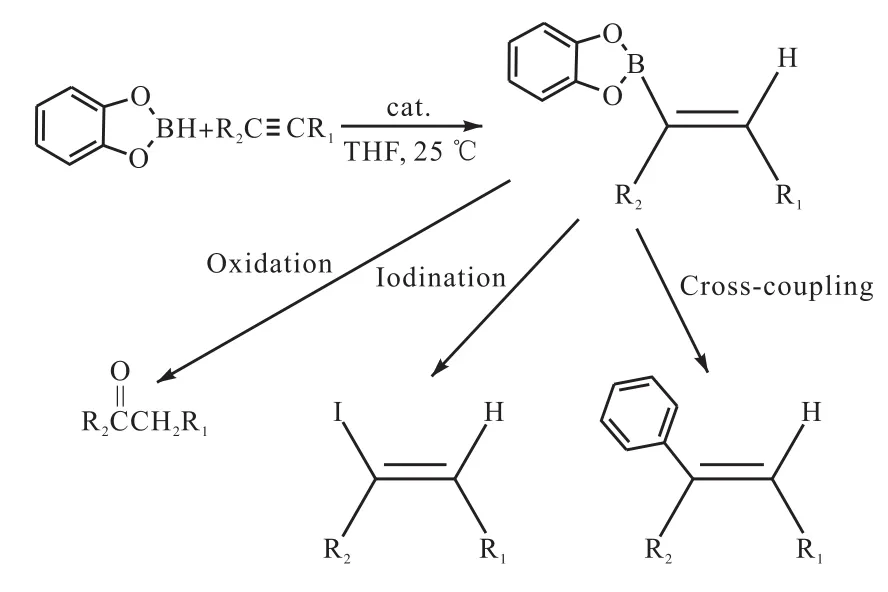
图17 炔烃硼氢化及其功能化产物Fig. 17 Hydroboration and functionalization of alkynes
Shirakawa等[42]以二环己基硼烷作为炔烃硼氢化反应的催化剂,反应可在室温条件下进行,反应转化率高并且对一系列的官能团具有耐受性.此外,由儿茶酚硼烷为硼氢化试剂制备的烯烃基儿茶酚硼烷在室温条件下与频哪醇进行反应制备频哪醇硼烷衍生物,由于频哪醇硼烷衍生物相对于儿茶酚硼烷衍生物更为稳定,从而被用来研究儿茶酚硼烷与烯烃或炔烃进行硼氢化反应之后所生成产物的顺反结构.
环戊二烯二甲基钛可以与过量的儿茶酚硼烷络合生成环戊二烯钛硼烷化合物,鉴于环戊二烯二甲基钛对烯烃硼氢化反应催化活性较高,Hartwig等[43]将此类络合物作为炔烃硼氢化反应在-20,℃条件下反应的催化剂,并对有无催化剂作用的反应结果进行了对比,结果发现催化剂和儿茶酚硼烷络合物在 40,℃的条件下会发生分解.不同环戊钛烯类催化剂对儿茶酚硼烷与己炔硼氢化反应过程当中插入点产物分布见表1[43].

表1 催化硼氢化产物分布Tab. 1 Distribution of catalyst hydroboration products
有机硼酸酯在催化不对称 1,4-加成反应过程当中扮演着重要的角色,但是由于其制备、分离、纯化较为困难使得有机硼酸酯在有机合成反应过程当中的应用受到限制.Takaya等[44]研究了 1-庚炔、苯乙炔、3,3-二甲基-1-丁炔、2-丁炔、3-甲氧基-1-丙炔在 0~70,℃的条件下与儿茶酚硼烷反应生成 2-烯基-1,3,2-苯并二氧硼烷衍生物.随后,顺式 1-庚烯基硼酸盐在催化剂的作用下与烯酮类化合物反应,烯烃基儿茶酚硼烷与烯酮类化合物当中的碳碳双键部分作用并插入到羰基间位的碳原子上(图18[44]).
Schafman等[45]对硼氢化反应过程中硼原子和氢原子在加成反应过程中的位阻效应进行了研究.在儿茶酚硼烷与 3-己炔进行硼氢化反应过程当中不加入任何自由基抑制剂和引发剂的条件下,反应产物当中80%,为3-己烯基-3-硼酸盐;当加入自由基抑制剂(加尔万氧基自由基)时,反应产物当中 65%,~75%,为反式硼氢化产物;当偶氮二异丁腈被加入到反应体系当中时,90%,的反应产物为反式硼氢化产物.顺式1-烯烃基硼酸也可以通过 1-氯-1-烯烃基硼酸酯在金属氢化物的作用下通过两步法制备,还可以通过1-烯烃基硼酸进行顺式硼氢化反应进行制备.

图18 炔烃硼氢化及烯酮加成反应Fig. 18 Hydroboration of 1-hexyne and allylation of alkylcatecholborane
Yang等[46]借助钯催化剂使得烷基硒乙炔与儿茶酚硼烷进行硼氢化反应生成顺式硼氢化产物 2-烷基硒-1-乙烯基儿茶酚硼烷,在碱性条件下 2-烷基硒-1-乙烯基儿茶酚硼烷与碘进行功能化反应生成 2-烷基硒-1-乙烯基碘,在镍类催化剂的作用下让格氏试剂与 2-烷基硒-1-乙烯基碘进行反应,从而将烯烃基团引入到分子当中.
苯乙炔与儿茶酚硼烷进行硼氢化反应生成两种烯烃基硼烷的异构体,末端炔烃在较高的温度下与儿茶酚硼烷进行顺式和反马氏加成反应的过程当中,催化剂对反应结果的影响并不是很明显,但是催化剂对硼氢化反应过程当中的化学选择性、区域选择性以及位阻选择性有较大的影响.
Ohmura等[40]首先开发了反式炔烃硼氢化产物的合成反应,他们发现顺式 1-烯烃基硼酸盐当中的 β氢原子并不是来自于硼烷试剂,而是反应过程中氘代末端炔烃的氘代氢选择性地转移到 β碳原子上.硼烷进行氧化加成反应的同时,硼氧键也会进行 1,2-迁移,经由还原性消除反应生成顺式 1-烯烃基硼酸盐.通过儿茶酚硼烷和频哪醇硼烷与炔烃进行催化硼氢化反应并对硼氢化产物分布进行总结,当过量的儿茶酚硼烷参与反应时,炔烃的顺式硼氢化反应将转换为反式硼氢化反应(图19[40]).
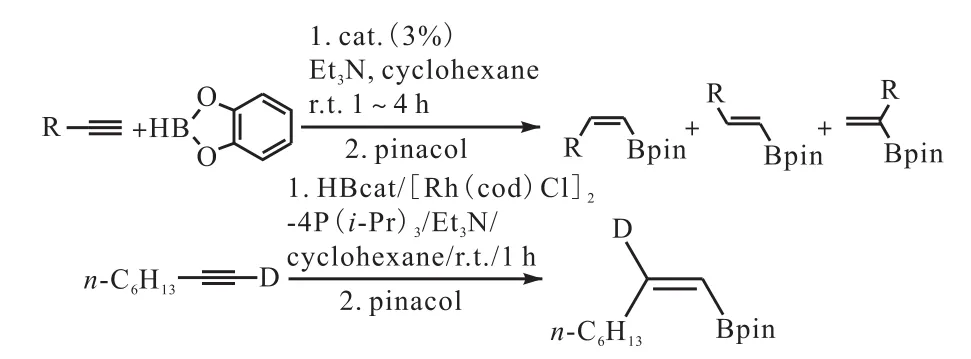
图19 炔烃硼氢化反应Fig. 19 Catalyzed hydroboration of alkynes
二氯化镍(NiCl2)可以使得儿茶酚硼烷在 70,℃的条件下与炔烃进行硼氢化反应合成重要的烯烃基儿茶酚硼烷中间体,反应产率和产物纯化程度均较高.Zaidlewicz等[12]将此类反应的应用范围进行了拓展,并对反应底物当中的官能团与儿茶酚硼烷在反应过程当中的耐受性进行了研究.在二氯化镍的作用下,末端炔烃与儿茶酚硼烷在室温条件下反应 3,h,反应产物在真空条件下的分离收率为 80%,~90%,,同时证实硼氢化插入点的区域选择性比没有催化剂作用条件下的要高(图20[12]).
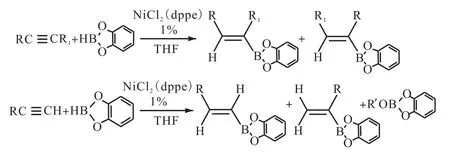
图20 炔烃类衍生物催化硼氢化反应Fig. 20 Hydroboration of functional derivatives of alkynes with catecholborane catalyzed by NiCl2at room temperature
近几年,金类催化剂已经被证明可用于活化不饱和碳碳键,此类催化剂不仅可以用来加速不饱和碳碳键的加氢反应,而且对其加氢反应的区域选择性和化学选择性有明显影响[47-49].Leyva等[50]借助于此类催化剂使得儿茶酚硼烷能够快速高效地在烯烃和炔烃混合物中选择性地进行硼氢化反应.
5 儿茶酚硼烷与其他不饱和官能团的硼氢化反应
Maki等[51]和Collum等[52]在1978年报道的羧酸与 2-羧基-1,3,2-苯并二氧环戊硼烷在儿茶酚硼烷的作用下生成儿茶酚硼烷衍生物中间体(图 21[51]),该中间体与等物质的量的胺进行亲核反应生成酰胺类化合物,作为拓展,将 2,3,4,5-四氯苯并二氧硼烷硼氢化产物产率与同样条件下 3,5-二三氟甲基苯基硼酸所引发反应的产物产率进行了研究.

图21 羧酸类化合物硼氢化及胺化反应Fig. 21 Carboxylic acids with amines using catecholborane as a condensing reagent and a possible catalytic pathway
儿茶酚硼烷所具有的路易斯酸特性使得其在四氯化钛和其他催化剂的作用下可与还原性试剂进行反应,Lindsley等[53]发现儿茶酚硼烷在 20,℃的二氯甲烷当中与苯乙酮完全反应需要 24,h,而将 10%,(物质的量)的钛类催化剂加入到体系当中后,反应在30,min内进行完全,将反应体系当中的沉淀物进行水解生成甲基苯甲醇.
Fujisawa等[54]使用恶唑硼烷衍生物作为儿茶酚硼烷在-90,℃条件下与三氟乙酰基联苯衍生物进行的反应的催化剂生成相应的醇类化合物(图22[54]).
Lebel等[55]利用铑类催化剂催化儿茶酚硼烷与脂肪醛进行硼氢化反应,随后将硼氢化产物进行氧化生成相应的醇类化合物.在铑类催化剂的作用下硼氢化转化率较高,反应体系中引入硼砂后反应转化率得到进一步提升.值得注意的是,反应过程中只有醛基参与反应,儿茶酚硼烷的化学选择性得以充分发挥.
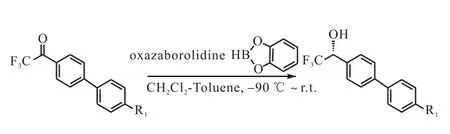
图22 羰基化合物硼氢化反应Fig. 22 Hydroboration of carbonyl derivates
Enders等[56]以恶唑硼烷类化合物作为催化剂,同时选用体积较大的儿茶酚硼烷作为硼氢化试剂与磺内酰胺进行反应以提高对映选择性,当过量的儿茶酚硼烷被加入到体系当中时,产物的对映选择性和产率都有所增加(图23[56]).
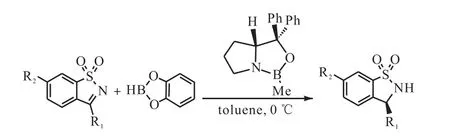
图23 手性磺内酰胺合成Fig. 23 Optimized reaction conditions for the enantioselective synthesis of chiral sultam
Zhang等[57]首先报道了手性磷酸催化剂,作为此类催化剂的应用范围的拓展.Enders等[58-59]将手性N-三氟甲磺酰基磷酰胺作为儿茶酚硼烷与酮亚胺类化合物反应的催化剂,儿茶酚硼烷与酮亚胺在低温条件下进行反应生成仲胺类化合物(图 24[58]),随后他们又在此基础上使用儿茶酚硼烷作为还原性试剂对α-酮酯类化合物选择性还原制备了α-羟基酯[60].
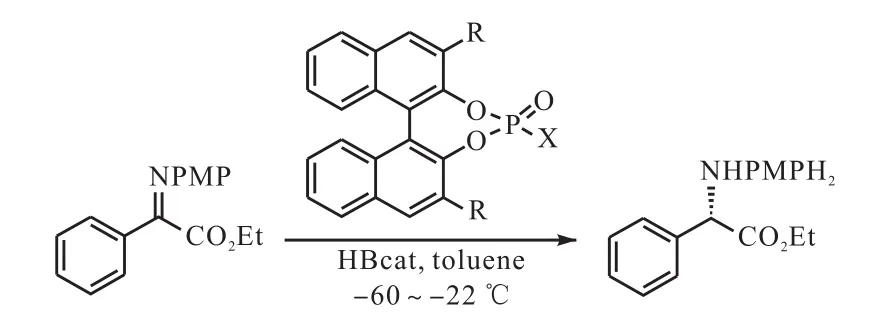
图24 甲氧苯基乙基亚胺硼氢化反应Fig. 24 Reduction of imino ester p-methoxyphenyl protected ethyl imino ester with catecholborane
亚砜类化合物脱氧生成硫醚类化合物在有机合成和生物化学反应过程当中扮演着重要的角色,然而许多亚砜还原反应所使用的还原性试剂较为昂贵并且难以进行处理,官能团耐受性较差,反应条件较为苛刻.Harrison等[61]将稳定易处理的儿茶酚硼烷作为亚砜类化合物的脱氧试剂,亚砜类化合物当中的官能团体积较大且为吸电子基团,因此需要加入过量的儿茶酚硼烷才能够使得亚砜类化合物当中的氧原子被完全去除(图25[61]).
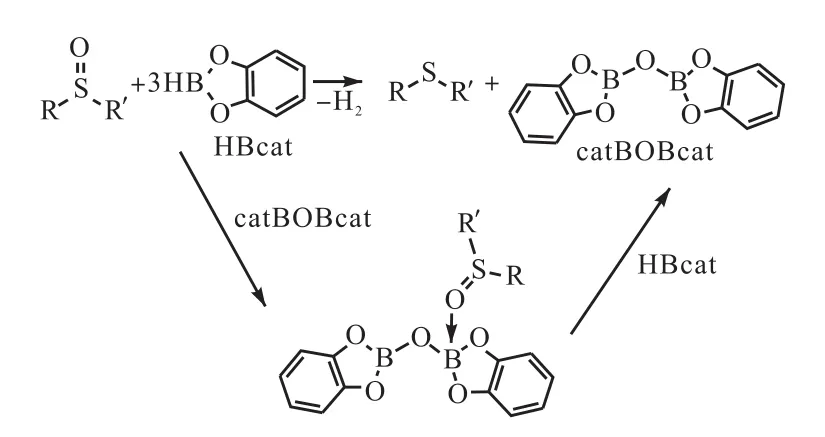
图25 亚砜类化合物硼氢化脱氧Fig. 25 Deoxygenation of sulfoxides using catecholborane
在没有催化剂作用的条件下,带有亚砜基官能团的化合物与儿茶酚硼烷完全反应,需要 500,h[61],Fernandes课题组[62-64]将 MoO2Cl2(H2O)2引入反应体系作为儿茶酚硼烷还原芳基亚砜类化合物的催化剂,在回流的四氢呋喃当中反应 0.5,h,硼氢化转化率即可达85%,以上.
作为一种比较新兴的硼氢化试剂,儿茶酚硼烷被Nakamura等[65]和 Khalimon等[66]应用在腈类物质的催化还原上,反应开始于通过 HBcat与催化剂形成硼氢化物中间体,在还原性消除后,形成硼氢化R-亚胺-儿茶酚硼酸盐作为单氢化产物,其进一步与HBcat反应,得到最终产品 R-N-二儿茶酚硼酸盐(图26[65]).
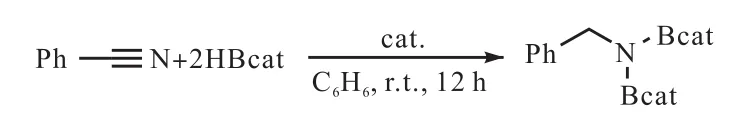
图26 苯甲腈硼氢化反应Fig. 26 Hydroboration of PhCN with HBcat
6 结 语
烷基儿茶酚硼烷是非常高效的自由基前驱体,它可以通过儿茶酚硼烷与烯烃或炔烃类化合物进行硼氢化反应获得.儿茶酚硼烷可以在氧气的诱导下高效地与烯酮和烯醇类化合物进行硼氢化反应,同时硼氢化过程当中出现的催化剂对其化学选择性产生影响,其他不饱和基团如羧基、羰基、酰胺和砜类基团同样可以在一定的反应条件下与儿茶酚硼烷进行硼氢化反应.
儿茶酚硼烷作为单H—B键硼烷,其硼氢化反应产物易于功能化和表征,这对拓展其应用范围产生了重要的影响,同时其商业化使得人们对其在各种反应当中应用研究变得更加方便.由于烷基儿茶酚硼烷易于被 TEMPO自由基氧化和易于被转化为其他基团,其在聚合物合成方面的应用也逐渐显现出来.烷基儿茶酚硼烷的 TEMPO氧化和碘取代化合物分别可以进行可逆加成-断裂链转移聚合反应[20-22]和链衰减转移自由基聚合反应[67-68],用于制备超支化聚合物和嵌段聚合物,这使得儿茶酚硼烷的应用从有机合成领域逐渐拓展到高分子合成领域.作为高分子领域应用的新尝试,由儿茶酚硼烷及其衍生物所制备的大分子引发剂,将对不同结构的聚合物的合成产生重要影响.
参考文献:
[1] Burgess K,Ohlmeyer M J. Transition-metal promoted hydroborations of alkenes,emerging methodology for organic transformations[J]. Chemical Reviews,1991,91(6):1179-1191.
[2] Beletskaya I,Pelter A. Hydroborations catalysed by transition metal complexes[J]. Tetrahedron,1997,53(14):4957-5026.
[3] Crudden C M,Edwards D. Catalytic asymmetric hydroboration:Recent advances and applications in carboncarbon bond-forming reactions[J]. European Journal of Organic Chemistry,2003(24):4695-4712.
[4] Vogels C M,Westcott S A. Recent advances in organic synthesis using transition metal-catalyzed hydroborations[J]. Current Organic Chemistry,2005,9(7):687-699.
[5] Newsom H C,Woods W G. Tributylin hydride reduction of chloroboranes[J]. Inorganic Chemistry,1968,7(1):177-178.
[6] Brown H C,Gupta S K. 1,3,2-Benzodioxaborole,a convenient monofunctional hydroborating agent. Simple new synthesis of alkaneboronic esters and acids from olefins via hydroboration[J]. Journal of the American Chemical Society,1971,93(7):1816-1818.
[7] Brown H C,Mandal A K,Kulkarni S U. Hydroboration.45. New,convenient preparations of representative borane reagents utilizing borane-methyl sulfide[J]. The Journal of Organic Chemistry,1977,42(8):1392-1398.
[8] Männig D,Nöth H. Contributions to the chemistry of boron. Part 160. A convenient synthesis of catecholatoborane and diborane[J]. Journal of the Chemical Society,Dalton Transactions,1985(8):1689-1692.
[9] Kanth J V B,Periasamy M,Brown H C. New economical,convenient procedures for the synthesis of catecholborane[J]. Organic Process Research & Development,2000,4(6):550-553.
[10] Braunschweig H,Guethlein F,Mailänder L,et al. Synthesis of catechol-,pinacol-,and neopentylglycolborane through the heterogeneous catalytic B-B hydrogenolysis of diboranes(4)[J]. Chemistry-A European Journal,2013,19(44):14831-14835.
[11] Männig D,Nöth H. Catalytic hydroboration with rhodium complexes[J]. Angewandte Chemie International Edition,1985,24(10):878-879.
[12] Zaidlewicz M,Melier J. Monohydroboration of conjugated dienes,alkynes and functionalized alkynes with catecholborane catalyzed by nickel(II)chloride and cobalt(II)chloride complexes with phosphines[J]. Main Group Metal Chemistry,2000,23(12):765-772.
[13] Vogels C M,Westcott S A. Recent advances in organic synthesis using transition metal-catalyzed hydroborations[J]. Current Organic Chemistry,2005,9(7):687-699.
[14] Vogels C M,Decken A,Westcott S A. Rhodium(I)acetylacetonato complexes containing phosphinoalkynes as catalysts for the hydroboration of vinylarenes[J]. Canadian Journal of Chemistry,2006,84(2):146-153.
[15] Lillo V,Mata J A,Segarra A M,et al. The active role of NHC ligands in platinum-mediated tandem hydroboration-cross coupling reactions[J]. Chemical Communications,2007(21):2184-2186.
[16] Vogels C M,O'Connor P E,Phillips T E,et al. Rhodiumcatalyzed hydroborations of allylamine and allylimines1[J]. Canadian Journal of Chemistry,2001,79(12):1898-1905.
[17] Brown H C,Chandrasekharan J. Hydroboration. 65.Relative reactivities of representative alkenes and alkynes toward hydroboration by catecholborane[J]. The Journal of Organic Chemistry,1983,48(25):5080-5082.
[18] Goddard J P,Le Gall T,Mioskowski C. Preparation of alcohols from alkenes via the homologation of boronates with(Trimethylsilyl)diazomethane[J]. Organic Letters,2000,2(10):1455-1456.
[19] Baban J A,Goodchild N J,Roberts B P. Electron spin resonance studies of radicals derived from 1,3,2-benzodioxaboroles[J]. Journal of the Chemical Society,Perkin Transactions 2,1986(1):157-161.
[20] Pouliot M,Renaud P,Schenk K,et al. Oxidation of catecholboron enolates with TEMPO[J]. Angewandte Chemie International Edition,2009,48(33):6037-6040.
[21] Uehara K,Wagner C B,Vogler T,et al. Poly(vinyl ketone)s by controlled boron group transfer polymerization(BGTP)[J]. Angewandte Chemie International Edition,2010,49(17):3073-3076.
[22] Wagner C B,Studer A. Synthesis of macro(alkoxyamines)via hydroboration of polyolefins with subsequent nitroxide oxidation for controlled NMP[J]. Macromolecular Chemistry and Physics,2010,211(23):2510-2516.
[23] Ollivier C,Chuard R,Renaud P. TEMPO-induced generation of alkyl radicals from B-alkylcatecholboranes[J]. Synlett,1999,6(6):807-809.
[24] Cadot C,Dalko P I,Cossy J,et al. Free-radical hydroxyllation reactions of alkylboronates[J]. The Journal of Organic Chemistry,2002,67(21):7193-7202.
[25] Wagner C B,Studer A. Synthesis of macro(alkoxyamines)via hydroboration of polyolefins with subsequent nitroxide oxidation for controlled NMP[J]. Macromolecular Chemistry and Physics,2010,211(23):2510-2516.
[26] Darmency V,Renaud P. Alkylation of ethyl pyruvate via reductive coupling of alkenes and Ethyl 2-(benzenesulfonylamino)acrylate[J]. CHIMIA International Journal for Chemistry,2005,59(3):109-110.
[27] Kumli E,Montermini F,Renaud P. Radical addition to 1,4-benzoquinones:Addition at O-versus C-atom[J].Organic Letters,2006,8(25):5861-5864.
[28] Beauseigneur A,Ericsson C,Renaud P,et al. B-alkylcatecholborane-mediated tandem radical conjugated addition-aldol cyclization[J]. Organic Letters,2009,11(16):3778-3781.
[29] Lüthy M ,Darmency V ,Renaud P. Modified B-alkylcatecholboranes as radical precursors[J]. European Journal of Organic Chemistry,2011,2011(3):547-552.
[30] Brown H C,Kabalka G W. Oxygen-induced reactions of organoboranes with the inert α,β-unsaturated carbonyl derivatives. Convenient new aldehyde and ketone synthesis via hydroboration[J]. Journal of the American Chemical Society,1970,92(3):714-716.
[31] Ollivier C,Renaud P. B-alkylcatecholboranes as a source of radicals for efficient conjugate additions to unsaturated ketones and aldehydes[J]. Chemistry-A European Journal,1999,5(5):1468-1473.
[32] Ollivier C,Renaud P. A convenient and general tin-free procedure for radical conjugate addition[J]. Angewandte Chemie International Edition,2000,39(5):925-928.
[33] Pozzi D,Scanlan E M,Renaud P. A mild radical procedure for the reduction of B-alkylcatecholboranes to alkanes[J]. Journal of the American Chemical Society,2005,127(41):14204-14205.
[34] Schaffner A P,Sarkunam K,Renaud P. Radical-mediated three-component coupling of alkenes[J]. Helvetica Chimica Acta,2006,89(10):2450-2461.
[35] Kapat A,König A,Montermini F,et al. A radical procedure for the anti-markovnikov hydroazidation of alkenes[J]. Journal of the American Chemical Society,2011,133(35):13890-13893.
[36] Schaffner A P,Renaud P. Tin-free radical allylation of B-alkylcatecholboranes[J]. Angewandte Chemie International Edition,2003,42(23):2658-2660.
[37] Davies A G,Roberts B P. Bimolecular homolytic substitution at a metal center[J]. Accounts of Chemical Research,1972,5(11):387-392.
[38] Garrett C E,Fu G C. Hydroboration of olefins with catecholborane at room temperature in the presence of N,N-dimethylacetamide[J]. The Journal of Organic Chemistry,1996,61(10):3224-3225.
[39] Brown H C,Gupta S K. Hydroboration. XXXIX. 1,3,2-Benzodioxaborole(catecholborane)as a new hydrobora tion reagent for alkenes and alkynes. General synthesis of alkane-and alkeneboronic acids and esters via hydroboration. Directive effects in the hydroboration of alkenes and alkynes with catecholborane[J]. Journal of the American Chemical Society,1975,97(18):5249-5255.
[40] Ohmura T,Yamamoto Y,Miyaura N. Rhodium- or iridium-catalyzed trans-hydroboration of terminal alkynes,giving(Z)-1-alkenylboron compounds[J]. Journal of the American Chemical Society,2000,122(20):4990-4991.
[41] Arase A,Hoshi M,Mijin A,et al. Dialkylboranecatalyzed hydroboration of alkynes with 1,3,2-benzodioxaborole in tetrahydrofuran[J]. Synthetic Communications,1995,25(13):1957-1962.
[42] Shirakawa K,Arase A,Hoshi M. Preparation of (E)-1-alkenylboronic acid pinacol esters via transfer of alkenyl group from boron to boron[J]. Synthesis,2004,2004(11):1814-1820.
[43] Hartwig J F,Muhoro C N. Mechanistic studies of titanocene-catalyzed alkene and alkyne hydroboration:Borane complexes as catalytic intermediates[J]. Organometallics,2000,19(1):30-38.
[44] Takaya Y,Ogasawara M,Hayashi T. Rhodium-catalyzed asymmetric 1,4-addition of 2-alkenyl-1,3,2-benzodioxaboroles to α,β-unsaturated ketones[J]. Tetrahedron Letters,1998,39(46):8479-8482.
[45] Schafman B,Matteson D S. Hydroboration of 3-hexyne:Free radicals with catecholborane but stereospecific addition of dichloroborane[J]. Main Group Metal Chemistry,1996,19(11):705-710.
[46] Yang D Y,Huang X. Regio-and stereoselective conversion of terminal alkylselenoacetylenes into(E)-2-alkylseleno-1-vinylic iodides via hydroboration-iodination[J]. Synthetic Communications,1996,26(24):4617-4621.
[47] Corma A,González-Arellano C,Iglesias M,et al. Gold nanoparticles and gold(III)complexes as general and selective hydrosilylation catalysts[J]. Angewandte Chemie International Edition,2007,46(41):7820-7822.
[48] Conte M,Davies T,Carley A F,et al. Selective formation of chloroethane by the hydrochlorination of ethene using zinc catalysts[J]. Journal of Catalysis,2007,252(1):23-29.
[49] Mizushima E,Sato K,Hayashi T,et al. Highly efficient Au(I)-Catalyzed hydration of alkynes[J]. Angewandte Chemie,2002,114(23):4745-4747.
[50] Leyva A,Zhang X,Corma A. Chemoselective hydroboration of alkynes vs. alkenes over gold catalysts[J].Chemical Communications,2009(33):4947-4949.
[51] Maki T,Ishihara K,Yamamoto H. 4,5,6,7-Tetrachlorobenzo [d] [1,3,2] dioxaborol-2-ol as an effective catalyst for the amide condensation of sterically demanding carboxylic acids[J]. Organic Letters,2006,8(7):1431-1434.
[52] Collum D B,Chen S C,Ganem B. A new synthesis of amides and macrocyclic lactams[J]. The Journal of Organic Chemistry,1978,43(22):4393-4394.
[53] Lindsley C W,DiMare M. Metal alkoxide catalysis of catecholborane and borane reductions. Mechanistic Studies[J]. Tetrahedron Letters,1994,35(29):5141-5144.
[54] Fujisawa T,Onogawa Y,Shimizu M. Structural effects of the oxazaborolidine derived from L-threonine in the reduction of(trifluoroacetyl)biphenyl derivatives with catecholborane[J]. Tetrahedron Letters,1998,39(33):6019-6022.
[55] Lebel H ,Ladjel C. Rhodium-catalyzed cascade reactions:A methylenation-hydroboration homologative process[J]. Journal of Organometallic Chemistry,2005,690(23):5198-5205.
[56] Enders D,Seppelt M. Catalytic enantioselective synthesis of 3-substituted benzosultams via Corey-Bakshi-Shibata reduction of cyclic N-sulfonylimines[J]. Synlett,2011(3):402-404.
[57] Zhang Z,Jain P,Antilla J C. Asymmetric reduction of ketones by phosphoric acid derived catalysts[J]. Angewandte Chemie International Edition,2011,50(46):10961-10964.
[58] Enders D,Rembiak A,Seppelt M. Asymmetric organocatalytic reduction of ketimines with catecholborane employing a N-triflyl phosphoramide Brønsted acid as catalyst[J]. Tetrahedron Letters,2013,54(6):470-473.
[59] Enders D,Rembiak A,Stoeckel B A. Chemo- and enantioselective Brønsted acid-catalyzed reduction of α-imino esters with catecholborane[J]. Advanced Synthesis &Catalysis,2013,355(10):1937-1942.
[60] Enders D ,Stöckel B A ,Rembiak A. Enantio-and chemoselective Brønsted-acid/Mg(n Bu)2catalysed reduction of α-keto esters with catecholborane[J]. Chemical Communications,2014,50(34):4489-4491.
[61] Harrison D J,Tam N C,Vogels C M,et al. A gentle and efficient route for the deoxygenation of sulfoxides using catecholborane(HBcat;cat=1,2-O2C6H4)[J]. Tetrahedron Letters,2004,45(46):8493-8496.
[62] Fernandes A C,Romao C C. Reduction of sulfoxides with boranes catalyzed by MoO2Cl2[J]. Tetrahedron Letters,2007,48(52):9176-9179.
[63] Cabrita I,Sousa S C A,Fernandes A C. Reduction of sulfoxides catalyzed by oxo-complexes[J]. Tetrahedron Letters,2010,51(47):6132-6135.
[64] Fernandes A C,Fernandes J A,Romão C C,et al. Highly efficient reduction of sulfoxides with the system borane/oxo-rhenium complexes[J]. Organometallics,2010,29(21):5517-5525.
[65] Nakamura G,Nakajima Y,Matsumoto K,et al. Nitrile hydroboration reactions catalysed by simple nickel salts,bis(acetylacetonato)nickel(Ⅱ)and its derivatives[J].Catalysis Science & Technology,2017(15):3196-3199.
[66] Khalimon A Y,Farha P M,Nikonov G I. Imido-hydrido complexes of Mo(Ⅳ):Catalysis and mechanistic aspects of hydroboration reactions[J]. Dalton Transactions,2015,44(43):18945-18956.
[67] Xu F,Dong P,Cui K,et al. New synthetic strategy targeting well-defined α,ω-telechelic polymethylenes with hetero bi-/tri-functionalities via polyhomologation of ylides initiated by new organic boranes based on catecholborane and post functionalization[J]. RSC Advances,2016,6(74):69828-69835.
[68] Wang H Y,Xu F,Cui K,et al. Synthesis of polymethylene-b-poly(vinyl acetate)block copolymer via visible light induced radical polymerization and its application[J]. RSC Advances,2017,7(67):42484-42490.
