基于位置信息熵的局部敏感哈希聚类方法
2018-04-18徐彭娜江育娥
徐彭娜 魏 静 林 劼 江育娥
(福建师范大学数学与信息学院 福建 福州 350108)
0 引 言
生物序列的聚类对生物信息学研究具有重要意义,聚类方法在生物序列分析和研究中受到了广泛的重视和应用。随着信息技术的发展,基因测序技术愈发成熟,生物数据的数量急剧增长,传统的方法对于海量的数据处理分析存在很多问题。数据挖掘技术是一种能从大量数据中提取有用的、潜在的有效信息的技术[1]。数据挖掘中的聚类能将具有某些相同特征的序列聚集在一起,从已知的功能、结构的序列探索出未知序列的有效信息,具有重要的意义。
由于生物序列数量的持续性增长,以Trapnell 等[2]为代表的K-中心点聚类算法将具有相似表达趋势的基因序列分为一组。当所有类簇的中心点都不改变时算法收敛,该算法在小型数据集上运行良好。然而由于两两比对进行聚类的方式在对大量数据聚类时存在计算量大、耗时久等问题[3]。因此在相似序列查找[4]和序列聚类上都提出用非比对方法解决两两比对存在的问题。以Bao等[5]为代表的非比对序列聚类算法无需对序列进行两两比对。虽然在效率上有一定程度的提高,但存在挖掘结果的可解释性不足和准确率偏离实际生物意义的问题,并不能完全满足生物学研究的需要。
针对以上问题,本文使用p稳定分布局部敏感哈希方法降低查找相似序列的时间复杂度;利用位置信息熵作为哈希函数的特征向量提高准确率;并在评估聚类结果时使用编辑距离作为距离度量以增强生物学可解释性,在此基础上提出基于位置信息熵的局部敏感哈希聚类方法LSH-E。该方法使用基于位置信息的标准熵作为局部敏感哈希函数的特征向量对生物序列进行聚类,其算法执行时间和数据集合大小成线性相关,对不同量级的数据集都有很好的实验结果,其在模拟数据和真实数据的实验结果均验证了该算法的有效性。
1 相关工作研究
在生物信息学中,聚类分析可以用于识别未知类别的生物序列所属的类别,揭示序列之间相互关系,进而分析生物序列功能和生物序列进化关系等[6]。
传统聚类算法如基于划分的K-medoid算法[7]、基于层次的全连接算法[8]等通过对序列进行两两比对来聚类,时间复杂度较高,因此传统算法不适用于海量数据的聚类分析。K-means算法需要确定聚类个数,序列数据的中心也不易计算,初始聚类中心具有随机性使得聚类结果不稳定,应用到生物序列中聚类效果不佳。基于BAG图[9]的聚类算法的结果虽然有效,但在类的分割时需要使用聚类单元引导,而基因库中的序列数目过多,导致其使用无向图表示过多的序列异常困难。Solovyov等[10]提出一种基于词频的高流通量中心聚类算法Afcluster,算法使用K词统计生物序列词频,基于词频信息确定中心使用k-means聚类。算法无需对序列进行两两比对,大大降低了时间复杂度,但在大数据量的情况下,随着类个数的增加,该算法的效率降低且存在聚类迭代时收敛缓慢的情况。Comin等[11]提出一种基于质量评价的非比对序列聚类算法Qcluster,由于聚类是基于序列的质量,该算法在指定聚类个数相对确定的情况下具有较高的准确度。但时间复杂度较高且运行时间不稳定,在数据量大且最终类个数不明确的情况下,存在着聚类时间过长或无法得到聚类结果的问题。Bao等[5]提出一种基于海明距离的空间块聚类算法Seed,该算法通过识别虚拟中心序列以及查找所有满足相似性参数的邻近序列来进行聚类。该算法的时间复杂度和空间复杂度和序列数成线性相关,在数量大的生物序列集合也能在较短时间得到聚类结果。但基于海明距离的生物序列聚类结果的可解释性和准确率偏低,可能偏离实际生物意义,不能完全满足生物学研究的需要。Fu等[12]提出一种基于并行的聚类算法CD-HIT,以减少序列冗余来应对快速增长的下一代产生的测序数据量。测试表明并行化到24个核心以及最多达8个核心的实验效果最佳,但其单机运行的时间效率偏低,对于边界比较不清晰的大数据集聚类效果差。
综上所述,为了解决现有算法存在时间效率不高、准确率较低、聚类结果的生物意义不明显的问题,本文提出LSH-E算法。使用标准熵记录生物序列的K词位置信息,应用编辑距离度量每对序列之间的距离,提高聚类分析的准确性以及增强其实际的生物意义。
2 理论基础
2.1 K词
近年来,在生物序列建模的数据预处理中对生物序列使用固定长度的滑动窗口获得K词集合,在K词的频数、概率统计等方法的基础上建立模型。生物序列的K词集合包含了频数和位置信息,体现了生物序列的特征和信息。由统计[13]可知,生物序列中的基因表达稳定的前提是一个K词的频繁出现,如果一个K词出现的极少,可能表示基因复制、基因表达被扰乱。在目前基于K词建立的模型中,多数只是基于K词的频数,对K词出现的位置信息未做处理,但是基因的重排、插入、转换和颠换等记载着大量的位置信息。生物序列的位置信息与生物序列的相似性研究有着重要的意义,本文使用信息熵标记K词的位置信息。
2.2 信息熵
Shannon 借鉴了热力学的概念将排除信息冗余的平均信息量称为信息熵[14],信息熵表示随机变量的概率分布函数,解决了信息分布度量化问题。信息熵满足对称性、非负性、确定性、扩展性、可加性及极值性等特性。设集合Y={x1,x2,…,xi,…,xn},i∈[1,n],假定xi∈Y的概率pi=P(xi),则Y的信息熵可定义为:
(1)
式(1)是信息熵公式,可以将其应用到生物序列上计算生物序列的信息熵[14]。其中集合Y可以表示为生物序列中K词的Locality Frequency集合,pi可以表示为每个K词的离散概率P的第i个离散概率。
2.3 局部敏感哈希
局部敏感哈希LSH基本是对样本数据进行映射变换后的,使得在原始空间的相邻点仍然保持大概率相邻,而在原始空间的不相邻点仍然保持大概率不相邻[15]。LSH主要应用于海量数据的检索、查找相似数据等。
局部敏感哈希簇需要满足两个条件:
(1) 如果d(x,y)≤d1,则f(x)=f(y)的概率至少为p1;
(2) 如果d(x,y)≥d2,则f(x)=f(y)的概率至多为p2。
其中x,y表示样本数据,d(x,y)表示x、y之间的距离,d1
3 基于位置信息熵的局部敏感哈希聚类方法(LSH-E)
由于生物数据的急剧增长,现有的大部分聚类算法存在着如时间复杂度高,无法对大量数据进行聚类,聚类准确率低,聚类结果的可解释性差等问题。为了解决时间复杂度高的问题,本文使用p稳定分布局部敏感哈希方法降低查找相似序列的时间复杂度。为了解决准确率低的问题,使用位置信息熵作为哈希函数的特征向量,利用位置信息来提高聚类的准确率。对于聚类结果的可解释性存在偏离实际生物意义的现象,本文在评估聚类结果时使用编辑距离作为距离度量。由于编辑距离被定义为将序列A映射到序列B所需操作({insert,delete,replace})的个数[16],这与生物序列基因的重组、遗传和变异等自然演变的实际生物意义相符。
3.1 基于信息位置的标准熵
对于一个给定的序列S=S[1,2,…,N],假如其字符集合为∑,S[i]表示在序列中的某个字符;S[i…j] 表示序列的一个子字符串;一个K词即指在序列S中的一个长度为k的一个子字符串。对于一个长度为N的序列S来说,其k的取值范围从1到N。 当k=1时,有N个长度为1的K词;当k=N时,有1个长度为N的K词(即S本身)。因此,对于序列S来说,总共有N(N+1)/2 个K词(k取值范围为[1,N])。
对生物序列使用固定长度K(1≤K≤N)的滑动窗口获得待处理K词集合Y,即预处理字长度为K。K词对生物序列符号使用长度为K的不重复生物序列集合,即K词集合包含|∑|K个特征词,其中生物序列符号集合为∑,|∑|是序列符号集合大小。统计每个特征词出现的位置及频数,使用式(2)计算每条序列的局部频率值LF值。
(2)
LF是通过依次计算每条生物序列的每个特征词出现的相邻位置差的倒数。在式(2)中,Y表示待处理K词集合,t表示同一个待处理K词出现的位置顺序,WtY表示待处理K词Y的第t次出现在生物序列的位置,LFtY表示待处理K词Y的第t个LF,z表示当前待处理K词频数。
使用式(3)、式(4)计算每个特征词的LF值的部分和ps、离散概率p。
(3)
(4)
式中:N为ps的个数。
使用式(5)计算每个特征词的熵值,使用式(6)对熵值进行标准化处理。
(5)
(6)
3.2 基于p-稳定分布的局部敏感哈希
基于 p-稳定分布的局部敏感哈希,其原理是利用一种随机化局部哈希方式对样本进行降维,使在高维相近的两个点在降维之后仍然大概率相近。该算法的具体过程是将样本投影到随机的直线上,对于随机直线而言,两两之间的方向矢量的分量服从p-稳定分布且互相独立。由于p-稳定分布的特性,两个样本的欧式距离与其投影距离是同分布的,因此两个样本的投影距离越小则表明其欧氏距离也就越小。将随机直线按等比例划分为线段,然后对各线段进行编号,则投影样本所在的线段编号值作为哈希函数的取值。该算法的哈希函数族可以表示为:
ha,b(v)=⎣(a·v+b)/w」
(7)
式中:v为d维的特征向量,a为与特征向量v个数相同的0到1之间的符合p-稳定分布的向量,b为0到w的任一整数,w为任意正整数,这样哈希函数ha,b(v)将一个d维空间向量v映射为一个整数。
3.3 基于位置信息熵的局部敏感哈希聚类方法
已知序列集合S={s1,s2,…,sn},对每条序列进行K词处理,根据式(2)至式(6)计算每条序列的基于位置信息的标准熵,得到序列位置信息熵集合E={e1,e2,…,e|∑|}。根据式(7)构造哈希个数为num_h的基于 p-稳定分布的局部敏感哈希集合H={h1,h2,…,hnum_h},将位置信息熵E作为哈希函数的特征向量,计算特征矩阵。将特征矩阵均分为r个行条,每个行条中哈希值相同的序列为同一个聚类,得到聚类个数为num_c的集合ClusterSet={cs1,cs2,…,csc,… ,csnum_c}。
LSH-E算法描述如算法1所示。步骤4)至步骤11)计算序列S的标准熵E,将序列转化为标准熵矩阵E表示,其中步骤6)、步骤7)、步骤8)分别使用式(2)、式(3)、式(4)计算,步骤9)使用式(5)和式(6)计算;步骤12)将标准熵作为特征向量,对其使用num_h个哈希计算得到哈希矩阵HM;步骤13)将HM均分为r个行条,每个行条中包含num_h/r行,将同一个行条的列拼接得到拼接哈希矩阵PHM;步骤15)至19)为聚类过程,其中步骤16)表示将至少有一个拼接值相同的序列聚为一类,步骤17)表示将已经被聚类的序列排除。
算法1LSH-E 算法描述
输入:序列集合S,哈希函数个数num_h,行条数r。
输出:聚类集合ClusterSet。
1)for(s∈S)
2) y=Kmer(s)
//对s进行K词处理得到Y
3) T=getT(Y)
//对K词结果计算频数并记录位置集合T
4) for(y∈Y)
5) if(Ty.length<=1){LFy=-1}
6) else{LFy=calculateLF(y)}
//计算LF值
7) pst=sum(LFy)
//计算部分和
8) p(t)= pst/ sum(ps)
//计算离散概率
9) e=Standardization(entropy)
//计算熵并对其标准化
10) end for
11)end for
12)HM=hashs(E, num_h)
//对特征矩阵进行分行条
13)PHM=PasteHashMatrix(HM , r)
//对特征矩阵进行拼接
14)i=1, Clustered=null
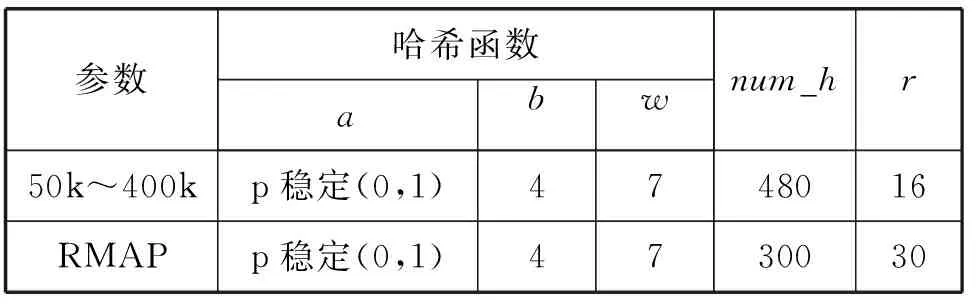
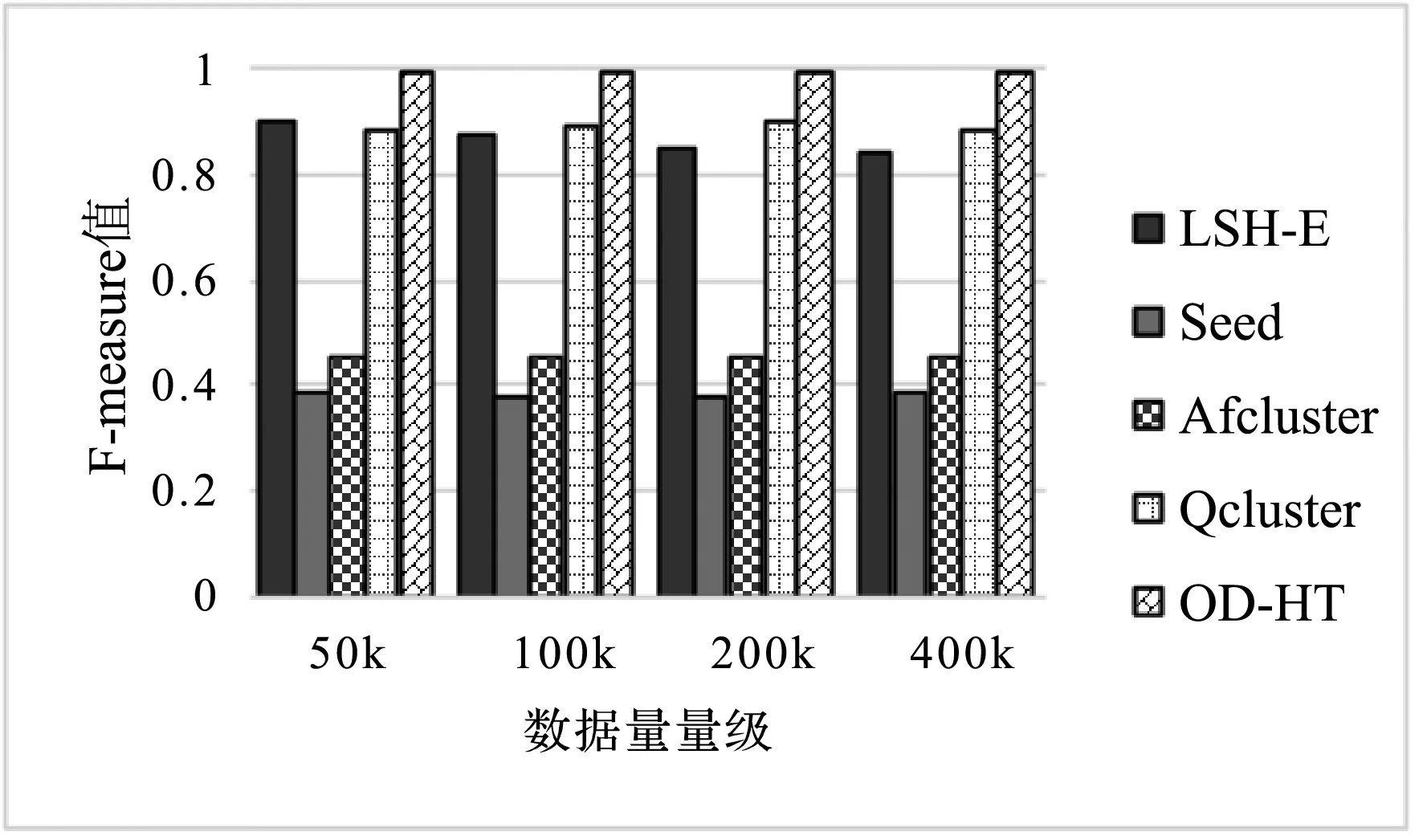
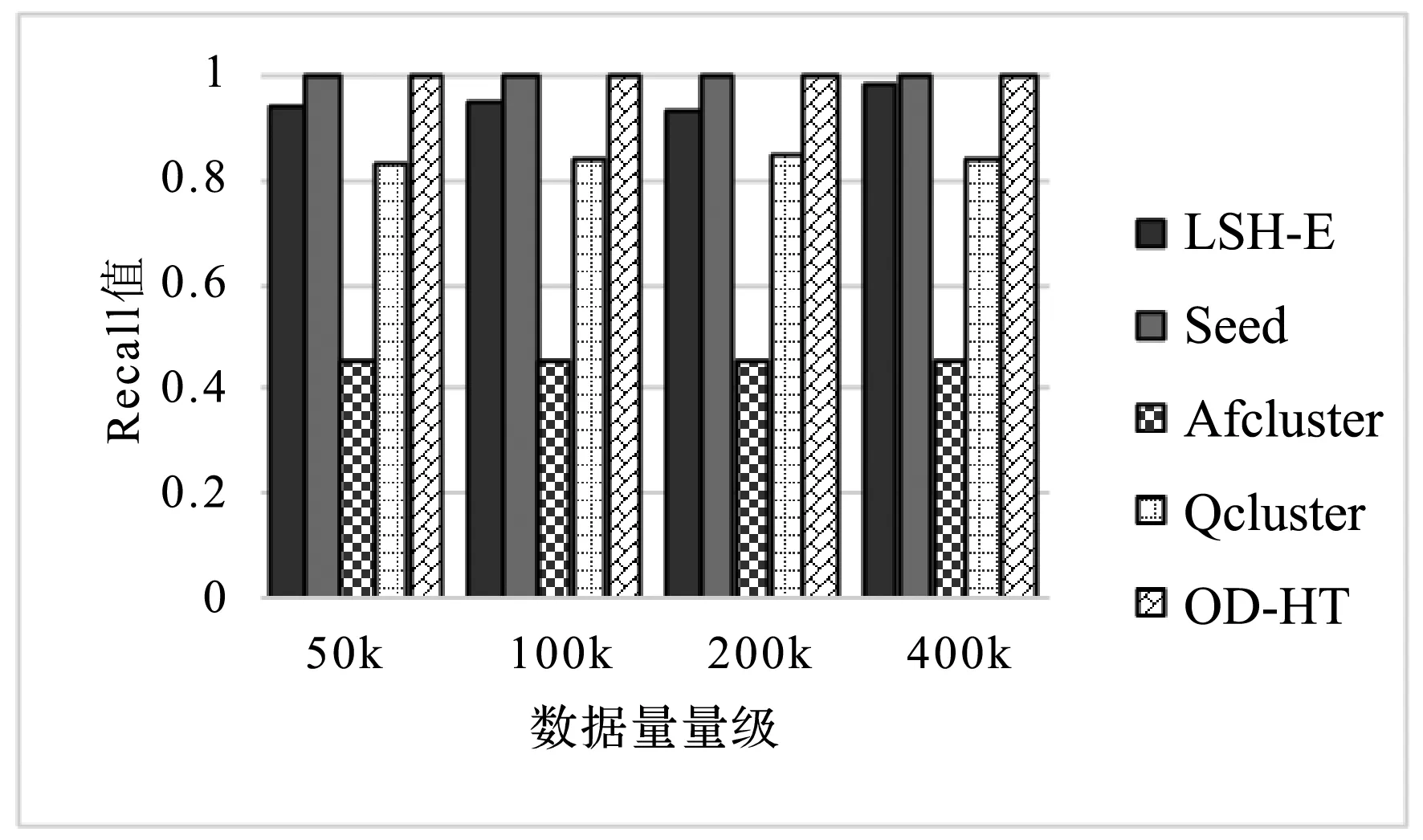
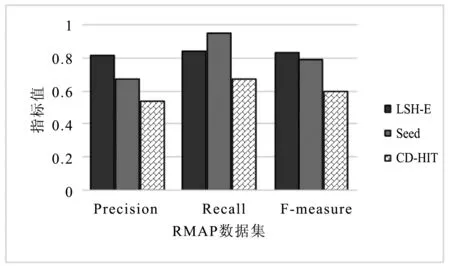
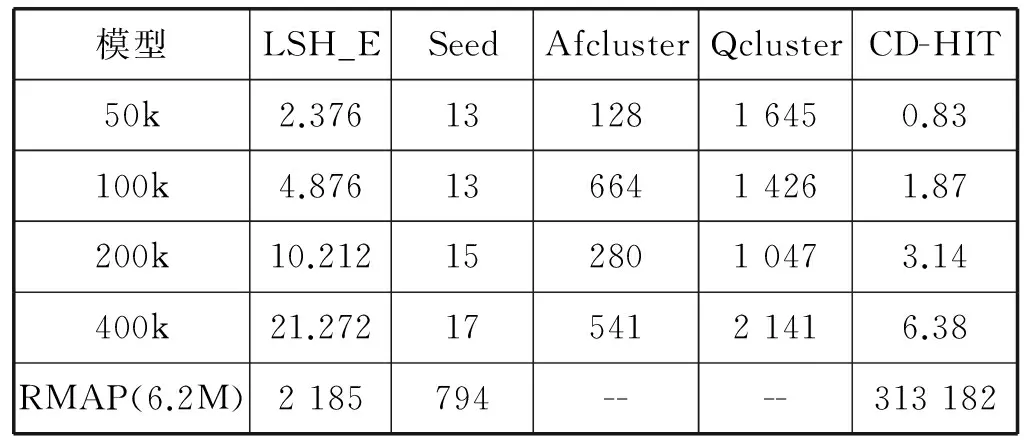
15)while(Clustered.length 16) temp=equal(PHM[,i]) 17) csc=removeClustered(temp , Clustered) 18) if(i∈Clustered){i++} 19)end while 20)return ClusterSet 实验使用两种数据集,真实数据集和模拟数据集。其中真实数据集RMAP[17]是二代测序数据集,每条序列的字符个数为47,该数据集中的总序列数为6 196 389,总体分布较均匀。模拟数据是先从真实数据集RMAP中选取编辑距离大于7的500条序列作为中心,再生成与每条中心编辑距离小于2的100条、200条、400条和800条序列,分别加上中心序列形成的50 500、100 500、200 500和400 500条 DNA生物序列集合,下文将其分别称为50k、100k、200k和400k。 LSH-E对DNA序列聚类的实验步骤如下: (1) 由于DNA序列中碱基集合为{A、C、G、T},|∑|=4,k=2,获得16个K词,对序列S进行K词模型映射处理,得到预处理字集合Y;(2) 根据式(2)、式(3)、式(4)计算每个特征词的LF值、部分和ps、离散概率p,本实验中对Y中只出现一次或未出现的K词的LF赋为-1;(3) 根据式(5)计算每个特征词的熵值,使用式(6)对熵值进行标准化处理;(4) 将熵值作为哈希函数簇的特征向量,计算DNA序列的特征矩阵;将特征矩阵进行分行条和拼接处理得到PHM矩阵;(5) 根据PHM计算聚类结果。 其中50k~400k、RMAP数据集的聚类实验参数如表1所示。 表1 LSH-E实验参数 表1中,a、b、w为式(7)中的参数,在实验中a为16维的0到1之间的符合p-稳定分布的向量,在不同w值的实验对比中发现,当w=7效果最佳;b选取0~7之间的中位数4;在不同num_h值的实验对比中发现对于50k~400k的数据,当num_h=480,r=16时,效果最佳,对于数据集RMAP,当num_h=300,r=30时,效果最佳。 算法Seed、 Qcluster、Afcluster、CD-HIT对DNA聚类的实验步骤如下: (1) 下载Seed(http://manuals.bioinformatics.ucr.edu/home/seed)、Qcluster(http://www.dei.unipd.it/~ciompin/main/qcluster.html)、Afcluster(https://github.com/luscinius/afcluster)和CD-HIT(https://github.com/weizhongli/cdhit)聚类方法的源代码,其中Seed源代码在linux系统中使用g++对源代码中的SEED.cpp文件进行编译,Qcluster、Afcluster和CD-HIT在linux系统中使用make进行编译操作;(2) 将DNA序列处理为实验需要的格式,其中Seed、Qcluster聚类实验需要.fastq格式的实验数据,由于RMAP数据集中的DNA数据无序列质量信息,将所有的序列质量信息置为相同的值,Afcluster、CD-HIT聚类实验直接使用.fasta格式的实验数据;(3) 对50k、100k、200k、400k和RMAP数据集计算Seed聚类结果,其中mismatch=3,shift=3,对50k、100k、200k、400k数据集计算Qcluster、Afcluster聚类结果,其中k值均设置为500,对50k、100k、200k、400k和RMAP数据集计算CD-HIT聚类结果,无需设置参数值。 实验采用精度P值(Precision值)、召回率R值(Recall值)、F值(F-measure值)、算法执行时间作为评估指标。由于真实数据集中没有明确的聚类结果,本文规定序列间的编辑距离小于等于3的序列真实为一个类。使用BCubed基准[18]对给定的数据集中的每条序列计算其精度和召回率,其评估方式如式(10)和式(11)所示。一条序列的精度表示用一个类中有多少个其他序列与该序列同属于一个类,一条序列的召回率表示有多少同一类的序列被分配在相同类中。 (8) 式中:(1≤i≤n)是实验结果的oi的类别, 是真实数据中oi的类别, 表示两个对象oi和oj(1≤i,j≤n,i≠j)在真实数据中的关系的正确性。 (9) (10) 使用式(11)计算F值。 (11) 对模拟数据和真实数据进行LSH-E实验,分别计算其F值、R值和P值,与Seed模型、Qcluster模型、Afcluster模型、CD-HIT模型的实验结果进行比较,其实验结果如图1-图4所示。 图1 五种不同方法得到的F值对比 图2 五种不同方法得到的R值对比 图3 五种不同方法得到的P值对比 图4 数据RMAP的不同方法的P值、R值和F值比较 图1是实验数据为50k、100k、200k和400k在五种方法(LSH_E、Seed、Afcluster、Qcluster、CD-HIT)下计算得出的F值。从图中可以看出,LSH_E模型所得到的F值明显大于Afcluster模型和Seed模型计算的到的F值,与Qcluster模型计算的F值相近。但 LSH_E模型得到的R值结果优于Qcluster模型(见图2),在时间效率上比Qcluster模型高了近百倍,CD-HIT模型所得的F值略优于LSH-E模型的F值。 图2是实验数据为50k、100k、200k、400k在五种方法(LSH_E、Seed、Afcluster、Qcluster、CD-HIT)下计算的R值。从图中可以看出,LSH_E模型所得到的R值明显大于Afcluster模型,优于Qcluster模型计算的到的R值。虽然LSH_E模型所得到的R值略小于Seed模型计算的R值,但LSH_E模型得到的P值结果显著优于Seed模型的P值(如图3所示),CD-HIT模型所得的R值略优于LSH-E模型的R值。 图3是实验数据为50k、100k、200k、400k在五种方法(LSH_E、Seed、Afcluster、Qcluster、CD-HIT)下计算的P值。从图中可以看出,LSH_E模型所得到的P值明显大于Seed模型和Afcluster模型计算的到的P值,LSH_E模型所得到的P值略小于Qcluster模型计算的P值,CD-HIT模型所得的P值略优于LSH-E模型的P值。 图4是实验数据为RMAP在LSH_E、Seed模型下计算的P、R、F值,Afcluster模型和Qcluster模型在实验数据为真实数据RMAP时得不到聚类结果。从图中可以看出,LSH_E模型所得到的P值和F值均优于Seed模型,LSH_E模型所得到的P值、R值和F值均优于CD-HIT模型约20%。在Afcluster模型和Qcluster下对RMAP数据聚类,由于算法执行时间过长(>24 h),未得到实验结果。 为了更详细地展示实验结果效率,本文统计了LSH_E、Seed、Afcluster、Qcluster和CD-HIT五种模型聚类时间作对比,时间对比如表2所示。 表2LSH_E、Seed、Afcluster、Qcluster、CD-HIT聚类时间 秒 由表2可知,在50k~400k的实验结果中,LSH-E的算法执行时间明显低于Afcluster和Qcluster的算法执行时间;在50k、100k、200k的实验中,LSH-E的算法执行时间少于Seed的算法执行时间,在400k、RMAP的实验中,LSH-E的算法执行时间高于Seed的算法执行时间;在50k~400k的实验中,LSH-E的算法执行时间略高于CD-HIT的算法执行时间,在RMAP的实验中,LSH-E的算法执行时间显著少于CD-HIT的算法执行时间。 在50k~400k数据中,LSH-E实验的P值、R值和F值均高于Seed模型和Afcluster模型。LSH-E模型的R值高于Qcluster模型,P值和F值略低于Qcluster模型。LSH-E实验的P值、R值和F值均低于CD-HIT模型,说明CD-HIT适合边界比较清晰的聚类情况。在RMAP真实数据中,Afcluster模型和Qcluster模型均无法计算结果,LSH-E模型的P值和F值均高于Seed模型,LSH-E模型的P值、R值和F值均高出CD-HIT模型约20%。由实验结果可知,LSH-E模型在两种数据集合均取得很好的实验结果,具有更高的时间效率和更好的生物解释性。 本文对DNA序列数据采用了基于位置信息熵的局部敏感哈希聚类方法进行聚类。使用长度为K的滑动窗口得到待处理K词集合和位置,计算K词的信息熵并对其标准化,作为局部敏感哈希簇的特征向量计算特征矩阵进行聚类,计算其P值、R值、F-measure值、和算法执行时间,对实验结果进行评估。使用模拟数据和真实数据集合进行LSH-E聚类,与Seed聚类、Qcluster聚类、Afcluster聚类结果进行对比。与Seed模型相比,LSH-E实验结果的具有更好的实验结果,在RMAP数据集的聚类中,LSH-E的F值较Seed的高出4.16%;与Qcluster模型相比,LSH-E在损失较小精度的情况下,在时间上是Qcluster模型1/100;与Afcluster模型相比,LSH-E模型无论在时间效率还是P值、R值、F值均得到了很大的提升;与CD-HIT模型相比,LSH-E模型在边界比较清晰的聚类中结果稍逊于CD-HIT模型,但在RMAP数据集的聚类中,LSH-E模型在时间上是CD-HIT模型的1/150,F值较CD-HIT的高出23.17%。实验结果表明,LSH-E在聚类时间上具有较大提高,随着数据集的增大,LSH-E同样取得很好的实验效果,且更具有生物解释性和实际意义。 [1] 宋建林. K-means聚类算法的改进研究[D]. 安徽大学, 2016. [2] Trapnell C, Cacchiarelli D, Grimsby J, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells[J]. Nature Biotechnology, 2014,32(4):381. [3] 周洋. 基因表达谱数据聚类分析的研究[D]. 咸阳: 西北农林科技大学, 2014. [4] Lin J, Adjeroh D A, Jiang B H, et al. K2: Efficient alignment-free sequence similarity measurement using the Kendall statistic[C]//IEEE International Conference on Bioinformatics and Biomedicine. IEEE, 2017:1128-1132. [5] Bao E, Jiang T, Kaloshian I, et al. SEED: efficient clustering of next-generation sequences[J].Bioinformatics, 2011,27(18):2502. [6] 杨恒宇. 生物序列数据挖掘技术研究[J]. 合肥工业大学学报自然科学版, 2012,35(9):1212-1216. [7] 戴伟. 基于MapReduce的K-Medoids并行算法研究[D]. 辽宁工程技术大学, 2015. [8] 熊赟. 生物序列模式挖掘与聚类研究[D].复旦大学, 2007. [9] 陈荣安. 基于改进的Bag-of-Features模型的图像分类研究[D]. 兰州大学,2015. [10] Solovyov A, Lipkin W I. Centroid based clustering of high throughput sequencing reads based on n-mer counts[J].Bmc Bioinformatics, 2013,14(1):268. [11] Comin M, Leoni A, Schimd M. Clustering of reads with alignment-free measures and quality values[J].Algorithms for Molecular Biology Amb, 2015,10(1):1-10. [12] Fu L, Niu B, Zhu Z, et al. CD-HIT: accelerated for clustering the next-generation sequencing data[J]. Bioinformatics, 2012, 28(23):3150. [13] 邓伟. 生物序列的相似性分析及k词模型研究[D]. 山东大学, 2015. [14] Bao J, Yuan R, Bao Z. An improved alignment-free model for dna sequence similarity metric[J]. BMC Bioinformatics, 2014,15(1):321. [15] 郑奇斌, 刁兴春, 曹建军,等. 结合局部敏感哈希的k近邻数据填补算法[J]. 计算机应用, 2016, 36(2):397-401. [16] 张勇. 基于高通量转录组测序的序列比对算法研究[D]. 中国科学技术大学, 2016. [17] Yang X, Chockalingam S P, Aluru S. A survey of error-correction methods for next-generation sequencing[J].Briefings in Bioinformatics, 2013,14(1):56. [18] Han J W, Kamber M. 数据挖掘:概念与技术[M].范明,孟小峰,译.3版.北京: 机械工业出版社,2007:315-319.4 实验与结果分析
4.1 数据来源
4.2 实验步骤
4.3 评测指标

4.4 实验结果及分析

5 结 语
