Identification of the impurities in o-chlorophenyl cyclopentyl ketone samples by high performance liquid chromatography-hybrid ion trap/time-of-flight mass spectrometry and preparation of o-chlorophenyl cyclopentyl ketone standard
2018-04-02MAXiaomengJINLanLIYaningZHENGHuiWEIYun000900038
MA Xiaomeng, JIN Lan, LI Yaning, ZHENG Hui, WEI Yun*(. , , , 0009, ; . , , , 00038, )
Impurities produced during synthesis and purification generally indicate the synthetic pathway of the chemical of interest [5]. Determining the impurities ino-chlorophenyl cyclopentyl ketone samples acquired from illegal sources may allow the manufacturers of the samples to be identified.o-Chlorophenyl cyclopentyl ketone has two common synthetic routes [6,7]. The impurities in an illicit sample could allow the synthetic route used to produce the sample to be determined and then the illicit manufacturer of theo-chlorophenyl cyclopentyl ketone to be identified. It is important to criminal investigations that impurities ino-chlorophenyl cyclopentyl ketone can be identified [8].
Identifying impurities is never easy. Structural similarities between a target product and an unknown impurity mean the product and impurity may behave similarly and have similar spectroscopic signatures, making it difficult to identify the impurity. High performance liquid chromatography-mass spectrometry (HPLC-MS) is a good technique for the identification impurities in drugs owe to its high sensitivity and selectivity. Many studies of impurities in drugs using HPLC-MS have been conducted in recent years [9-19]. Several studies focused on identifying impurities in illegal drugs have been published [20,21]. Khajeamiri et al. [20] analyzed impurities in illicit methamphetamine samples seized in Iran by GC-MS and HPLC-MS. They found that 1,2-dimethyl-3-phenylaziridine was the most common impurity. It indicated that most of the methamphetamine samples were synthesized using pseudoephedrine as a starting material. However, the studies mentioned above provide limited data, and few impurities in illegal drugs have been identified. With the development of mass spectrometry technology, high resolution MS (HRMS), which can provide accurate mass, has been used for prediction formulas and elucidation structural of impurities in drugs. Among these, high performance liquid chromatography-hybrid ion trap-time of flight mass spectrometry (HPLC-IT/TOF MS) has emerged as a desirable tool to structurally characterize impurities. The reason is HPLC-IT/TOF MS has specific capacity for providing both the MSnability of the IT and the high resolution as well as mass accuracy capabilities of the TOF. Accurate data obtained by HPLC-IT/TOF MS allows studies of impurities in illegal drugs to be performed. Li et al. [22] identified 10 impurities in the illegal drug 2,5-dimethoxy-4-ethylphenethylamine (2C-E) from the MSnspectra and the elemental compositions of the impurities acquired by HPLC-IT/TOF MS, and this allowed the 2C-E synthetic route to be identified.
Unlike 2C-E,o-chlorophenyl cyclopentyl ketone is a different type of illicit drug, little information of the manufacture makes criminal investigations difficult. In order to identify the manufacturing source ofo-chlorophenyl cyclopentyl ketone, a useful method to determine the impurities ofo-chlorophenyl cyclopentyl ketone samples is necessary. The aim of this study was to develop an HPLC-IT/TOF MS method for the identification of unknown impurities ino-chlorophenyl cyclopentyl ketone. The structures and fragmentation pathways of the impurities were fully elucidated using the hybrid mass measurements. A method for the separation ofo-chlorophenyl cyclopentyl ketone from confiscated drugs was also established.
1 Experimental
1.1 Chemicals and reagents
Acetonitrile, formic acid, and methanol (all HPLC grade) were purchased from Fisher Scientific (Fair Lawn, NJ, USA). Trifluoroacetic acid (HPLC grade) was obtained from Dima Technology (Beijing, China). Sodium hydroxide (analytical grade) was purchased from Beijing Chemical Factory (Beijing, China). Ultrapure water was prepared using a Synergy Purification System (Millipore, Molsheim, France). Illicito-chlorophenyl cyclopentyl ketone samples seized in cases and reference standards ofo-chlorophenyl cyclopentyl ketone (1 g/L) were provided by the Chinese Institute of Forensic Science, Ministry of Public Security (Beijing, China).
1.2 Sample preparation
The illicito-chlorophenyl cyclopentyl ketone sample was dissolved in HPLC grade methanol to give a mass concentration of 1 g/L, and the solution was placed in an ultrasonic bath for 10 min. The solution was filtered through a 0.22 μm filter and then stored at 4 ℃ until undertaking HPLC-IT/TOF MS and analytical HPLC analyses.
For the preparative HPLC procedure, the illicito-chlorophenyl cyclopentyl ketone sample was dissolved in HPLC grade methanol to give a mass concentration of 20 g/L, and the solution was placed in an ultrasonic bath for 10 min. The solution was filtered through a 0.22 μm filter and then stored at 4 ℃ until use.
1.3 Instrument analysis method
1.3.1HPLC-IT/TOF MS conditions
An HPLC-IT/TOF MS instrument (Shimadzu, Kyoto, Japan) and HPLC-MS Solution software were used. HPLC procedures were conducted using a Shimadzu HPLC system consisting of an LC-20 AD solvent delivery pump, an SIL-20 AC autosampler, a DGU-20A3 degasser, an SPD-M20A photodiode array detector, a CBM-20A communication base module, and a CTO-20A column oven. Separation was carried out on an Inertsil ODS-SP column (150 mm×4.6 mm, 5 μm, GL Sciences, Tokyo, Japan) using mobile phase A of 0.05% (v/v) formic acid aqueous solution and mobile phase B of methanol with gradient elution. Binary gradient elution was performed using a linear gradient from 40%B to 82%B between 0 and 35 min at a flow rate of 1 mL/min. The split ratio for the MS system was 1∶0.3. The sample chamber in the autosampler was kept at 4 ℃, and the column temperature was kept at 40 ℃. The sample injection volume was 5 μL. The detection wavelength was set at 209 nm.
A hybrid IT/TOF MS instrument (Shimadzu, Japan) with an electrospray ionization (ESI) source, used in positive and negative ion modes, were used for the identification of the impurities in the illicito-chlorophenyl cyclopentyl ketone samples. The positive and negative electrospray voltages were +4.5 and -3.5 kV, respectively. The total effluent was transferred directly from detector to the hybrid IT/TOF MS instrument without splitting. MS analyses were performed in full scan mode over anm/zrange of 100-2 000, and data-dependent MSnwas performed on the suspected impurity ions. The nebulizing gas flow rate was 1.5 L/min. The curved desolvation line and heat block temperatures were both 200 ℃. The ion source temperature was set as 120 ℃. The detector voltage was 1.85 kV. The TOF and IT region pressures were 1.8×10-4and 1.8×10-2Pa, respectively. The ion accumulation time was 30 ms, and the precursor ion isolation width was anm/zof 3. Ultra-high-purity argon was used as both the cooling and the collision gas in collision-induced dissociation (CID) experiments, and the normalized collision energy was set at 50% for MS2and 60% for MS3.
External mass calibrations over them/zratio range 100-2 000 were performed using a 2.5 mmol/L trifluoroacetic acid sodium solution. The infusion pump flow rate was 5 mL/min. Tuning was performed in autotuning mode, and the tuning results were saved as a tuning file.
1.3.2Analytical HPLC
The analyses were performed on a Shimadzu LC-20AD HPLC system equipped with a SIL-20A auto-injector and a SPD-M20A diode array detector (DAD) set at 209 nm. Separation was achieved using a Shim-pack VP-ODS column (250 mm×4.6 mm, 5 μm, Shimadzu) fitted with a C18 guard column. The column temperature was 40 ℃. The mobile phase was 70% (v/v) methanol aqueous solution containing 0.05% (v/v) trifluoroacetic acid, and the flow rate was 1.0 mL/min. The sample injection volume was 5 μL.
1.3.3Preparative HPLC
Preparative HPLC separation was performed with a Shimadzu (Japan) LC-8A preparative HPLC system equipped with a SPD-M20A DAD, an SIL-10AP automatic injector, and an FRC-10A automatic fraction collector. Separation was achieved using a Shim-pack VP-ODS preparative column (250mm×20 mm, 15 μm, Shimadzu, Japan). Methanol-water (85∶15, v/v) was used as the mobile phase. The flow rate was 8 mL/min. The detection wavelength was 209 nm. The injection volume was 1 mL. The preparative HPLC equipment was controlled by LC Solution Chromatography Data software (Shimadzu, Japan).
1.4 Identification of o-chlorophenyl cyclopentyl ketone
o-Chlorophenyl cyclopentyl ketone was identified in the preparative HPLC procedure by MS and1H-NMR spectrometry. MS were performed on an HPLC-IT/TOF MS instrument (Shimadzu, Japan) with a SPD-M20A photodiode array detector using the method described in Section 1.3.1.1H-NMR spectrometry was performed in CD3OD using an Avance DRX-500 (500 MHz) NMR spectrometer (Bruker Biospin, Germany) at room temperature. A pulse width of 4.2 μs and a pulse repetition time of 2 s were used.
2 Results and discussion
2.1 HPLC-IT/TOF MS analysis and the o-chlorophenyl cyclopentyl ketone fragmentation pathway
An HPLC-IT/TOF MS method for the identification of the fragments produced by the 1 g/Lo-chlorophenyl cyclopentyl ketone standard was established. Theo-chlorophenyl cyclopentyl ketone standard MSndata shown in Fig. 1a were acquired in positive ion mode. A protonatedo-chlorophenyl cyclopentyl ketone molecular ion [M+H]+was observed atm/z209.072 1, and was assigned the formula C12H14ClO+. MS2of the ion atm/z209.072 1 gave a product ion atm/z191.056 4, indicating that 18 Da had been lost. This represented the loss of H2O. A five-membered ring then opened and a neutral loss of C2H4was lost. MS3of the ion atm/z191.056 4 gave a fragment ion atm/z163.036 9. A MS2product ion atm/z139.006 4 formed through the loss of cyclopentyl from the ion atm/z209.072 1. MS3of the ion atm/z139.006 4 gave product ions atm/z111.036 7 and 77.019 8, which were attributed to the loss of CO and Cl from the ion atm/z139.006 4. The proposedo-chlorophenyl cyclopentyl ketone fragmentation pathway is shown in Fig. 1b.
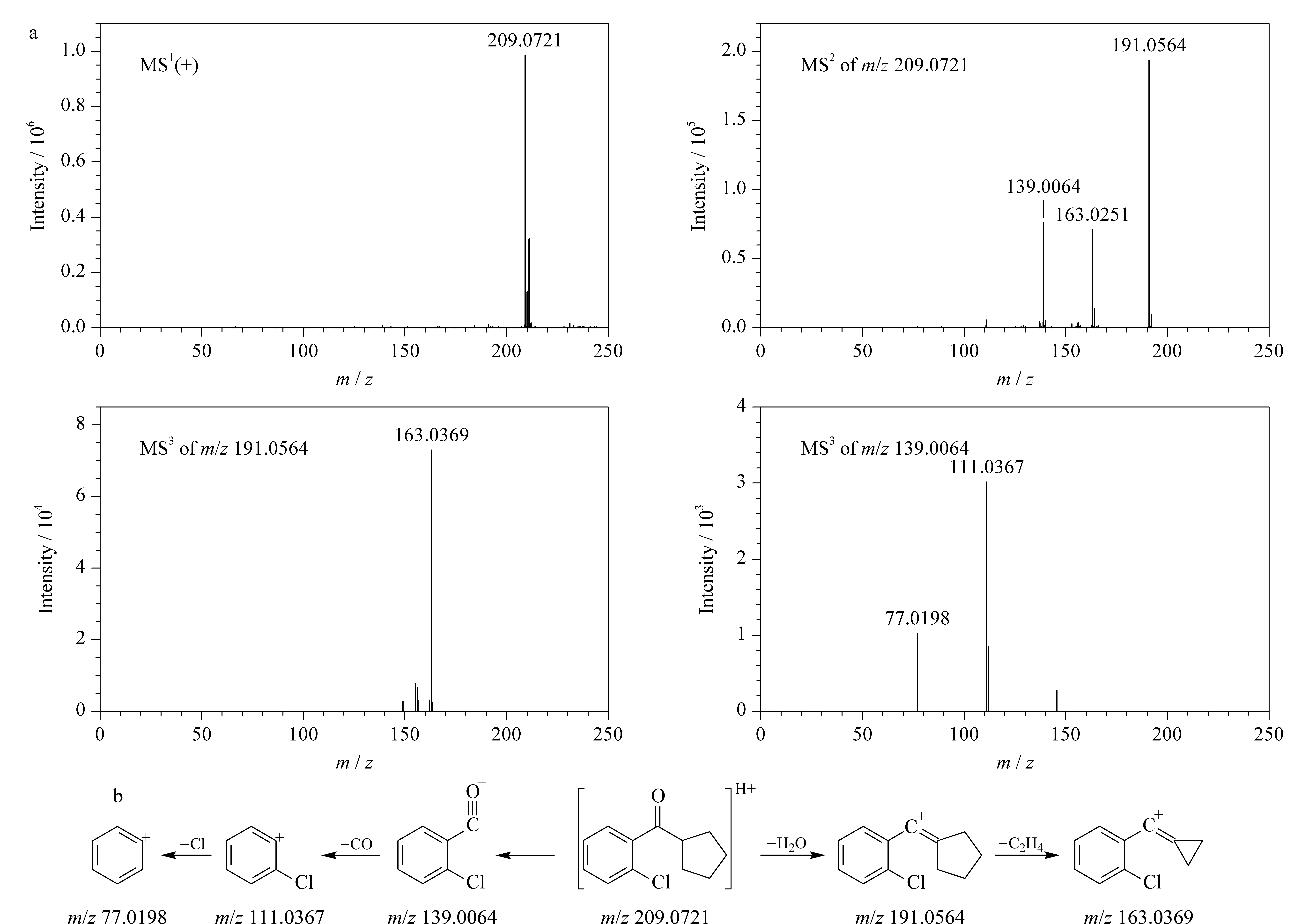
Fig. 1 (a) MSn spectra and (b) the proposed fragmentation pathways of o-chlorophenyl cyclopentyl ketone
2.2 Identification of impurities in the o-chlorophenyl cyclopentyl ketone samples by HPLC-IT/TOF MS
An HPLC-IT/TOF MS method for the identification of two unknown impurities in the 1 g/Lo-chlorophenyl cyclopentyl ketone samples seized in cases was established. This method was based on the method described above.

Fig. 2 (a) HPLC chromatogram and (b) TIC of an o-chlorophenyl cyclopentyl ketone sample The conditions were the same as those in section 1.3.1.
An HPLC chromatogram at 209 nm wavelength using DAD detector and total ion chromatogram (TIC) (Fig. 2a and 2b) were obtained using the HPLC-IT/TOF MS method described in section 1.3.1. As shown in Fig. 2b, two impurities were detected ino-chlorophenyl cyclopentyl ketone samples. The impurity characterization results are summarized in Table 1.
It can be seen from Fig. 2a and Table 1 that the major impurity 1, which had a molecular ion peak [M+H]+atm/z295.992 7, eluted at 8.392 min. Elemental analysis using the HPLC-MS Solution software was performed to determine the molecular formula of impurity 1. Several theoretically possible molecular formulae were found. Using the minimal difference method, the most likely molecular formula for impurity 1 was found to be C14H8Cl2O3. Seen from Fig. 3a, MS2analysis of the precursor ion atm/z295.992 7 gave a product ion atm/z139.000 6, which was assigned the formula C7H4OCl+. The MS3fragment ion atm/z111.002 9 formed through the neutral loss of CO from the ion atm/z139.000 6. The impurity 1 precursor ion atm/z295.992 7 losto-chlorobenzoic acid ion and CO (in Fig. 3a). Based on the exact mass data and similar fragmentation pathway ofo-chlorophenyl cyclopentyl ketone, we found that impurity 1 could beo-chlorobenzoic acid anhydride. All the fragments were similar to fragments in data provided by the National Institute of Advanced Industrial Science and Technology (Japan). The proposed impurity 1 fragmentation pathway is shown in Fig. 3b, and the structure of impurity 1 is shown in Fig. 3c.
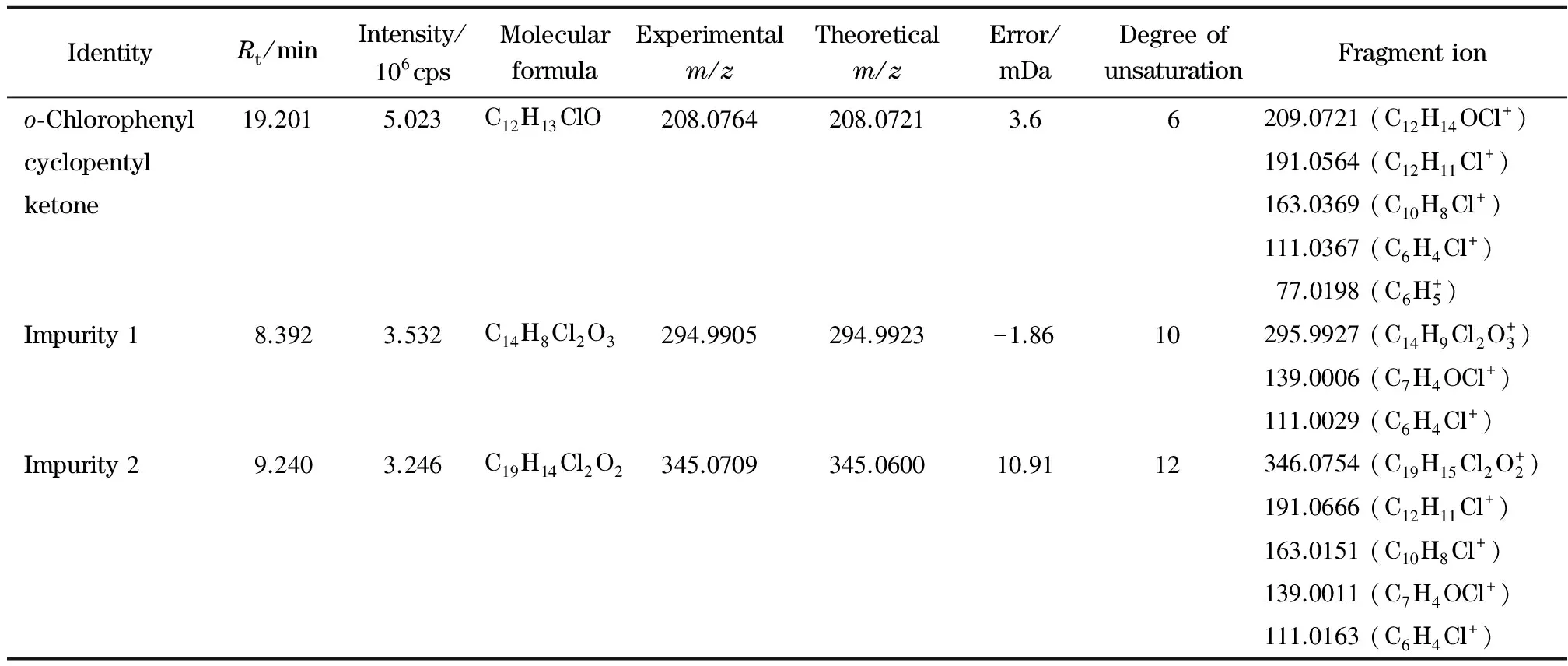
Table 1 Characterization summary of the impurities identified in o-chlorophenyl cyclopentyl ketone samples
Rt: retention time.

Fig. 3 (a) MSn spectra, (b) the fragmentation pathways and (c) the structure of the impurity 1
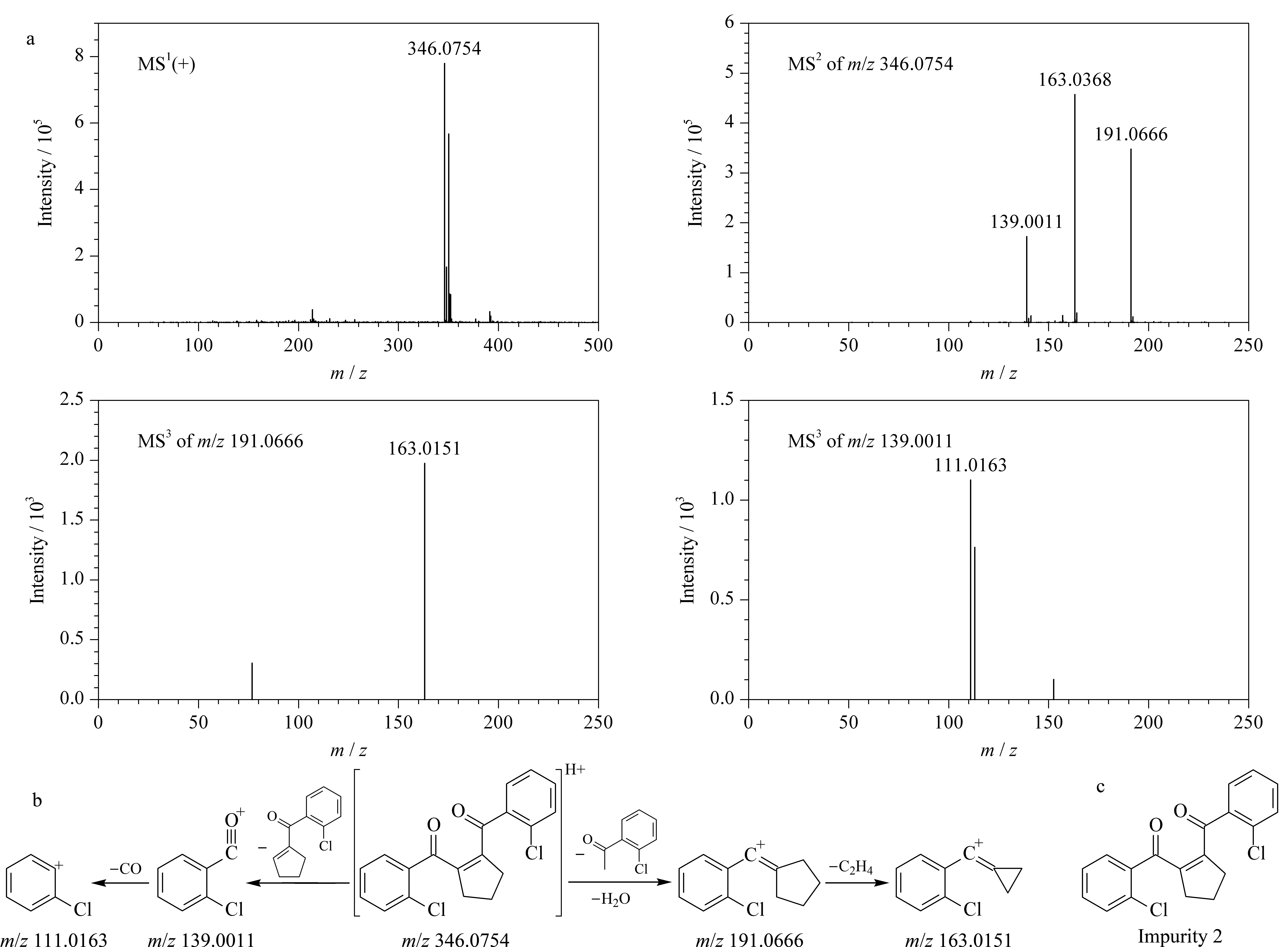
Fig. 4 (a) MSn spectra, (b) the fragmentation pathways and (c) the structure of the impurity 2

2.3 Synthetic route

Fig. 5 Synthetic methods of (a) o-chlorophenyl cyclopentyl ketone, (b) o-chlorobenzoic acid anhydride, and (c) 1,2-di-o-chlorobenzoylcyclopentene
There are many methods for synthesizingo-chlorophenyl cyclopentyl ketone. A route for the synthesis of theo-chlorophenyl cyclopentyl ketone in the samples that were analyzed was proposed based on the impurity data shown above. The synthetic route is shown in Fig. 5a, and was the same as that described by Zhang and Li [6].o-Chlorobenzoic acid anhydride is formed through the dehydration of twoo-chlorobenzoic acid molecules, using phosphorus pentachloride as a catalyst. The proposed synthetic route for the impurityo-chlorobenzoic acid anhydride is shown in Fig. 5b. 1,2-Di-o-chlorobenzoylcyclopentene appeared to be an adduct of the starting materialo-chlorobenzoyl chloride and cyclopentene. The reaction mechanism (Fig. 5c) involves the formation of ano-chlorobenzoyl carbocation fromo-chlorobenzoyl chloride, catalyzed by aluminum chloride. The next step is the electrophilic addition of cyclopentane, giving theo-chlorophenyl cyclopentyl ketone ion intermediate, which loses a proton to giveo-chlorophenyl cyclopentene. The addition ofo-chlorophenyl cyclopentene to theo-chlorobenzoyl carbocation occurs, then 1,2-di-o-chlorobenzoylcyclopentene is formed through the loss of a proton.
2.4 Preparative HPLC separation of o-chlorophenyl cyclopentyl ketone from the illicit samples
The 1 g/L illicito-chlorophenyl cyclopentyl ketone samples seized in cases were analyzed by HPLC-IT/TOF MS using the method described in Section 1.3.1. Seen from the TIC and the mass spectra shown in Fig. 1 and Fig. 2, theo-chlorophenyl cyclopentyl ketone samples containedo-chlorophenyl cyclopentyl ketone ([M+H]+m/z209.072 1), small amounts ofo-chlorobenzoic acid anhydride ([M+H]+m/z295.992 7), and 1,2-di-o-chlorobenzoylcyclopentene ([M+H]+m/z346.075 4).
The HPLC method used to analyze the 20 g/Lo-chlorophenyl cyclopentyl ketone samples seized in cases was established as described in Section 1.3.2.o-Chlorophenyl cyclopentyl ketone and the impurities absorbed UV light strongly at 209 nm and relatively strongly throughout the range of 190-500 nm. The wavelength used to detecto-chlorophenyl cyclopentyl ketone was 209 nm.o-Chlorophenyl cyclopentyl ketone was well separated from the other components (Fig. 6a). The eluent from preparative HPLC should be removed easily when the fraction was concentrated and dried by evaporator.o-Chlorophenyl cyclopentyl ketone could be separated from the other components in 56 min using the analytical HPLC method, which was relatively time-consuming. So the methanol-water containing 0.05%(v/v) trifluoroacetic acid (70∶30, v/v) in system was not suitable for preparative HPLC. The methanol-water (85∶15, v/v) described in Section 1.3.3 was used. The proportion of methanol was increased to 85% (v/v) to shorten the elution time, ando-chlorophenyl cyclopentyl ketone was able to be separated well from the other components within 40 min (Fig. 6b). In addition, when the injection volume was selected as 1 mL, it enabled a high preparation efficiency as well as purity. The preparedo-chlorophenyl cyclopentyl ketone standard was 99.53% pure, determined from the HPLC peak area percentages. An analytical HPLC chromatogram of theo-chlorophenyl cyclopentyl ketone fraction is shown in Fig. 6c.

Fig. 6 (a) Analytical HPLC chromatogram, (b) prepara-tive HPLC chromatogram of an o-chlorophenyl cyclopentyl ketone sample and (c) an analytical HPLC chromatogram of the o-chlorophenyl cyclopentyl ketone fraction from the preparative HPLC system
2.5 Structure identification
Theo-chlorophenyl cyclopentyl ketone fraction was freeze-dried and analyzed by MS and1H-NMR. The mass spectrum contained peaks atm/z209.063 1, 191.056 8, 163.029 8, 139.009 4, 111.007 0, and 77.019 8, indicating that the product waso-chlorophenyl cyclopentyl ketone. Combustion elemental analysis: calculated for C12H13ClO, C 69.73%, H 5.32%; found, C 69.33%, H 5.29%. The1H-NMR (CD3OD)δvalues were 1.66 (m, 4H), 1.86 (m, 4H), 3.84 (t, 1H), and 7.48 (m, 4H), the same as found in a previous study [23]. These data indicated that the compound waso-chlorophenyl cyclopentyl ketone.
3 Conclusions
A novel and effective HPLC-IT/TOF MS method for the identification of the impurities in the drugo-chlorophenyl cyclopentyl ketone was established. Ano-chlorophenyl cyclopentyl ketone fragmentation pathway was proposed. Two impurities were identified from their MSnspectra and elemental composition analysis results. Furthermore, the synthetic route of illicito-chlorophenyl cyclopentyl ketone samples was deduced on the basis of impurities analysis. The characteristic impurities could be used to identify the sources ofo-chlorophenyl cyclopentyl ketone samples seized by the authorities and to regulate the clandestine synthesis ofo-chlorophenyl cyclopentyl ketone. The results indicated that the proposed method will be very useful for the identification of impurities in other illegal drugs. Theo-chlorophenyl cyclopentyl ketone reference standard will allow reliable qualitative and quantitative analyses of illegal drugs.
[1]Menhart V, Mitinko V. Czech Patent, 277268. 1992-12-16
[2]Zheng H, Liu J, Mei Y J, et al. Catal Lett, 2012, 142(5): 573
[3]Wenger J, Winternitz P, Zeller M. Europe Patent, 0542685. 1993-05-19
[4]Zhou Z G, Wang Y. Journal of Yunnan Police Officer Academy, 2013(3): 24
[5]Lee J S, Han E Y, Lee S Y, et al. Forensic Sci Int, 2006, 161(2/3): 209
[6]Zhang L J, Li B Z. Chinese Journal of Medicinal Chemistry, 1995, 5(1): 47
[7]Stevens C L, Thuillier A, Daniher F A. United States Patent, 3254124. 1966-03-31
[8]Ko B J, Suh S, Suh Y J, et al. Forensic Sci Int, 2007, 170(2), 142
[9]Patil P, Vaidya I. Int J Pharm Res Sch, 2013, 2(2): 54
[10]Adouani I, Du M, Hang T J. Chromatographia, 2013, 76(9/10): 499
[11]Thommas S, Chandra Joshi S, Vir D, et al. J Pharmaceut Biomed, 2012, 63(3): 112
[12]Debremaeker D, Visky D, Chepkwony H K, et al. Rapid Commun Mass Sp, 2003, 17(4): 342
[13]Yuan Y Z, Zhang M, Fan X L, et al. J Pharmaceut Biomed, 2013, 80(3): 1
[14]Wang M, Li Y P, Wang Y, et al. J Pharmaceut Biomed, 2013, 84(84c): 69
[15]Liu L, Cao N, Ma X, et al. J Pharmaceut Biomed, 2016, 117: 325
[16]Kumar N, Devineni S R, Gajjala P R, et al. J Pharmaceut Biomed, 2016, 120(9): 248
[17]Dousa M, Srbek J, Rádl S, et al. J Pharmaceut Biomed, 2014, 94(3): 71
[18]Zhang H, Sun L, Zou L, et al. J Pharmaceut Biomed, 2016, 128: 18
[19]Kumar N, Devineni S R, Dubey S K, et al. J Pharmaceut Biomed, 2017, 137: 268
[20]Khajeamiri A R, Faizi M, Sohani F, et al. Forensic Sci Int, 2012, 217(1/3): 204
[21]Power J D, O’Brien J, Talbot B, et al. Forensic Sci Int, 2014, 241(8): e13
[22]Li Y N, Wang M C, Li A X, et al. Anal Methods, 2016, 8(46): 8179
[23]Zhang P, Li C H. Synthetic Commun, 1986, 16(8): 957
