毛细管电泳技术在单克隆抗体药物分析中的应用
2018-04-02陈泓序北京理工大学生命学院北京100081
陈泓序, 屈 锋(北京理工大学生命学院, 北京 100081)
随着单克隆抗体技术近40年的发展,其在生物医学研究和生物制药以及临床治疗中的应用发展迅速,已占有重要地位。单克隆抗体药物是通过基因工程生产的蛋白质药物,具有特异性高、作用机制明确、效果显著、经济效益大等优势,成为近年来生物医药产业的重要增长点。2017年全球销量排名前十位的处方药中有5个为单克隆抗体药物,艾伯维公司的阿达木单抗(Humira)更是连续6年蝉联全球最畅销药第一名。因欧美发达国家具有技术和投资优势,目前它们已垄断了大多数上市的单克隆抗体药物的生产。2020年,全球排名前6的单抗药物的专利均将到期[1],因此,目前国内外药企都掀起了生物类似药的研发热潮,以抢占单克隆抗体药物的巨大市场。为确保单抗产品的安全有效、质量可控,各国的药监部门都在不断地完善和提升生物类似药的质量控制要求[2]。我国《国家药品安全“十二五”规划》中也明确了药物一致性评价的质量要求,即仿制药品要与原研药质量和疗效一致。与小分子药物不同,抗体类药物不仅分子质量大,结构复杂,而且可经糖基化或其他翻译后修饰,导致其结构具有高度不均一性(异质性)。传统药物分析方法已无法满足对其纯度分析、等电点测定及电荷异质性分析和N-寡糖分析等需求。对单克隆抗体类药物的表征分析和质量控制提出了新的要求和挑战[3]。
毛细管电泳技术(capillary electrophoresis, CE)特别适合蛋白质等极性分子的分析,它具有分离效率高、模式多、分析速度快等优势。近年来已发展成为单克隆抗体药物多项表征中不可或缺的技术[4,5]。与传统分离技术如HPLC技术相比,CE在单抗药物的多项分析中有着独特的优势(见表1)。在CE的多种分离模式中,依靠相对分子质量大小分离的毛细管凝胶电泳(capillary gel electrophoresis, CGE)、依靠等电点分离的毛细管等电聚焦(capillary isoelectric focusing, CIEF)和依靠电荷/质量比分离的毛细管区带电泳(capillary zone electrophoresis, CZE)是单抗药分析中常见的3种模式。CGE用于还原和非还原单抗药物的纯度分析和N-寡糖分析,CIEF用于等电点及电荷异质性分析,CZE用于快速电荷异质性分析(见表2)。这些CE技术已经被全球大部分生物制药公司作为常规方法用于单克隆抗体药物的质量控制、药品生产质量管理规范(good manufacturing practice, GMP)条件下的产品放行和稳定性研究、配方开发中的过程表征和过程验证等环节。CE用于单克隆抗体药物的分析已被欧洲药典、美国药典等多国法规机构所认可。《中国药典》(2015版)三部单抗药物总论中也收录了毛细管电泳方法。本文综述了单克隆抗体药物质控和生产中的纯度分析、等电点及电荷异质性分析和N-寡糖分析的方法及应用进展。
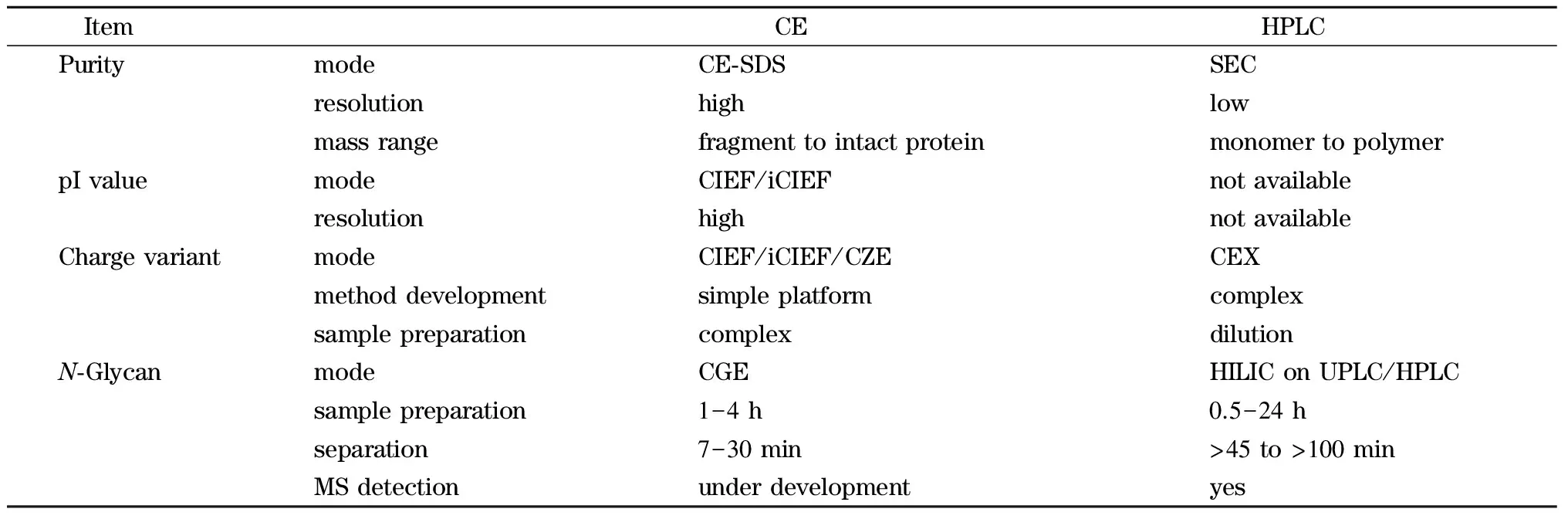
表 1 CE与HPLC技术在单克隆抗体药物分析中的比较Table 1 Comparison of CE and HPLC for therapeutic monoclonal antibody (mAb) analysis

表 2 单克隆抗体药物分析中的CE方法Table 2 CE methods for therapeutic mAb analysis
*PDA: photo-diode array; UV: ultraviolet-visible; LIF: laser-induced fluorescence.
1 单克隆抗体药物的纯度分析
传统的十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis, SDS-PACE)可对10~220 kDa范围内的变性蛋白质单体、聚合体及片段进行分离,是基于相对分子质量大小分离蛋白质的常规技术。在SDS-PAGE方法中,SDS与蛋白质分子具有特定的结合比(1.4 g SDS/1 g蛋白质)。蛋白质分子与SDS结合后变性而形成不同相对分子质量的带负电荷的SDS-蛋白质复合物,它们在电泳条件下沿电场的反方向泳动。因不同相对分子质量的SDS-蛋白质复合物在凝胶筛分介质中的缠绕和作用力不同而迁移速度不同,其迁移时间与相对分子质量的对数值成正比,进而可以估算蛋白质药物的相对分子质量。此外SDS-PAGE还用于表征蛋白质药物的一致性和纯度。常用的SDS-PAGE方法采用考马斯亮蓝染色法和银染法来检测蛋白质复合物。其中考马斯亮蓝法的检出限约为1 μg/mL,银染法灵敏度更高,检出限可达10 ng/mL。虽然SDS-PAGE是蛋白质分离中的常规技术,在实验室广泛使用,但其方法需要制胶、染色、脱色等繁琐的人工操作,且具有耗时长,重复性差,只能进行半定量等局限性。
1987年,Karger等[6]报道了使用交联聚丙烯酰胺作为筛分介质的毛细管凝胶电泳,将传统的平板凝胶转移到毛细管中进行。由于在毛细管中填入固态凝胶容易造成堵塞和气泡等问题。因此,1996年,Guttman等[7]开发了以线性聚合物取代交联聚丙烯酰胺的无胶筛分毛细管电泳。目前单抗分析中的毛细管凝胶电泳也早已不再使用交联的凝胶。由于样品蛋白质需要与SDS结合形成复合物,这种模式也被称为十二烷基硫酸钠无胶筛分毛细管电泳(CE-SDS-NGS),简称CE-SDS。CE-SDS方法的出现,解决了传统SDS-PAGE面临的问题,它具有明显的优势:(1)快速:30 min之内完成冲洗和分离过程;(2)自动化程度高:无需制胶、染色、脱色等人工操作,所有的冲洗、进样、分离及检测过程全部自动化完成;(3)分辨率高:商品化毛细管电泳仪可精确、有效控温,使分离电压可达30 kV,相比于最高几百伏电压的SDS-PAGE方法,分析速度加快,分辨率也有了极大的提高;(4)定量准确度高:根据待测蛋白质自身的紫外吸收或标记试剂的荧光信号强度实现准确定量,可避免SDS-PAGE检测中染料和蛋白质嵌合程度不同引起的差异;(5)在线检测:在毛细管出口端的窗口处进行光学检测,提高了方法的准确度。(6)重复性好:全自动的操作提高了分离的重复性。
CE-SDS方法相对传统SDS-PAGE的巨大优势使其成为生物制药行业中蛋白质药物纯度(大小异质性)分析的最优方法,逐渐被越来越多的国际知名制药企业所采用,也得到包括中国在内的多国药典的普遍认可[8]。目前大部分生物制药公司广泛采用贝克曼库尔特公司的商品化试剂盒进行CE-SDS分析。Lin等[9]对CE-SDS和SDS-PAGE进行单抗药物纯度分析的结果进行了比较,CE-SDS不仅在耐用性、精密性、线性、重复性和分辨率等方面均远远优于SDS-PAGE法,更重要的是它可以实现还原抗体的重链与非糖基化重链(non-glycosylated heavy chain, NGHC)的分离和准确定量(见图1)。因抗体药物的非糖基化重链直接影响药物的生物学功能,如抗体依赖的细胞毒性(antibody-dependent cellular cytotoxicity, ADCC)、半衰期、体内外稳定性等,因此非糖基化重链信息是抗体药物质控的关键指标,而CE-SDS方法是目前唯一能将抗体的非糖基化重链与重链分离、并能对非糖基化重链准确定量分析的技术,通过它能获得SDS-PAGE无法得到的抗体关键信息。

图 1 还原抗体药物的CE-SDS分析电泳图[9] Fig. 1 Electropherogram of mAb by reduced capillary electrophoresis-sodium dodecyl sulfate (CE-SDS)[9] Peak identifications: 1. internal standard (IS); 2. light chain (LC); 3. non-glycosylated heavy chain (NGHC); 4. heavy chain (HC).

图 2 (a)不同培养基下还原抗体及(b)不同克隆中非还原抗体的CE-SDS分析电泳图[11]Fig. 2 Electropherograms of (a) reduced CE-SDS for products from different cell culture and (b) non-reduced CE-SDS for products from different clone[11] The lowest trace in (a) was PNGase F-treated mAb produced in cell culture C.
强生公司的Zhang等[10]利用CE-SDS对单抗药表征的方法进行了系统的优化和方法验证,包括对还原和非还原抗体所用的分离条件及样品处理条件。还原抗体CE-SDS中,可采用巯基乙醇、二硫苏糖醇(DTT)或三(2-羧乙基)膦(TCEP)作为还原剂将单抗药的二硫键断裂,形成重链和轻链;在非还原抗体CE-SDS中,添加碘乙酰胺(IAM)或碘乙酸(IAA)封闭活性位点来减小蛋白质碎片的生成。默克公司的Rustandi等[11]报道了CE-SDS技术在单抗药物生产的各个环节中的广泛应用。它不仅可进行包括NGHC在内的产品相关杂质的定量分析,还可用于细胞培养和克隆筛选环节中对NGHC、抗体碎片和聚集体进行监控,来进行培养基的优化和克隆筛选(见图2)。图2a展示了用CE-SDS方法分析还原抗体来进行培养基优化的结果。相比于培养基A,培养基B产生的抗体中所含非糖基化重链含量更少,因此B被选为最佳的培养基。C培养基中产生了额外的峰(HC+2 kD),将C培养基中的样品经过肽-N-糖苷酶F(PNGase F)处理后的电泳图(最下面的电泳图)显示,HC+2 kD中含有额外的N-糖基。因非还原抗体中二硫键并未断裂,不应出现碎片峰,故分析结果常用来判断样品中的碎片杂质。例如,重链二聚体(P100,约100 kD)是克隆筛选中关注的重要杂质,而在体积排阻色谱(size-exclusion chromatography, SEC)中,P100和完整的抗体(P150,约150 kD)无法实现较好的分离。采用CE-SDS则可以对它们进行高效的分离。图2b展示了用CE-SDS方法分析非还原抗体来进行克隆筛选的结果。克隆3因为产生最少的P100而被选中进行后续的开发。此实验中,采用有效毛细管长度10 cm的快速分离模式,加快了分离速度,适合高通量的克隆筛选。另外,在产品稳定性研究中,同样可利用CE-SDS法分析不同条件下碎片杂质的生成情况,进而评估单抗药的稳定性。Li等[12]利用CE-SDS法对单抗药在不同存储条件下碎片的变化做了准确定量。在40 ℃下放置28天后,两个碎片峰的总含量由1.1%增加至4.6%。杂质片段的鉴定是结合SEC-HPLC和RPLC-MS方法进行的。经鉴定,该碎片为重链决定簇互补区(CDR)上Ser-Ser键断裂的产物。此外,王文波等[13]还使用还原和非还原抗体的CE-SDS法进行埃博拉复方抗体(包含3种单抗)的纯度分析。李萌等[14]使用CE-SDS方法对抗体偶联药物(antibody-drug conjugates, ADC)进行大小异质性分析。
目前CE-SDS用于单抗药的纯度分析主要采用两种检测手段,紫外检测(包含UV和PDA)和激光诱导荧光检测(laser-induced fluorescence, LIF)[15]。与紫外检测器相比,激光诱导荧光检测器灵敏度高,在抗体药物纯度分析中,可以检测到痕量的蛋白质杂质。而且,因激光诱导荧光检测法可以消除背景胶中紫外吸收引起的基线波动,使得分析方法的基线更加平整,便于减小不同操作者的积分误差,提高自动积分的准确性。基因泰克公司的Hunt等[16]最早报道了使用CE-SDS-LIF检测的方法。他们以5-羧基四甲基罗丹明作为染料对单克隆抗体药物进行标记,方法检出限可达9 ng/mL,与SDS-PAGE的银染法检测灵敏度相当。他们又进一步对该CE-SDS-LIF方法中的样品缓冲液pH值、染料和蛋白质样品的比例、反应温度等条件进行了考察和优化,提出在蛋白质样品与SDS预处理的过程中加入烷基化试剂,用于抑制非还原蛋白质在分离过程中蛋白质片段的形成。通过考查特异性、准确性、回收率、线性范围、检出限、精密性及重复性,对方法进行了详细的验证,进而将CE-SDS-LIF方法确定为基因泰克公司单抗药物质量控制的放行标准[17]。之后,Michels等[18]用3-(2-呋喃)-2-甲酰基喹啉(3-(2-furoyl)-quinoline-2-carboxaldehyde, FQ)染料标记蛋白质,使用CE-SDS-LIF方法进行单抗药物的纯度分析;染料FQ在氰基的作用下与蛋白质发生偶联反应,偶联之前FQ不具有荧光,而与蛋白质反应后才可发出荧光信号。因此标记过程中无需纯化除去过量的荧光染料,大大简化了蛋白质标记的过程。采用此法对单抗药的杂质定量分析,检出限达到10 ng/mL,可以检测到0.02%的杂质蛋白质[19]。
为保证CE-SDS方法的稳定和可靠,以安进公司为代表的抗体联盟,对CE-SDS方法进行了更深入的研究。他们发现如果凝胶产生气泡,则会引起电流中断等现象。为了解决这些问题,提高CE-SDS方法的重复性,他们提出了采用反吸法移取凝胶溶液和电泳过程中在毛细管两端同时施加相同的压力的方法[20],他们研究的这些技巧使CE-SDS方法更加稳定,为其在生物制药行业的广泛应用奠定了重要基础。为了评估CE-SDS方法作为单抗药物质控标准的可行性,惠氏、安进、礼来、基因泰克、辉瑞等13家欧美制药企业[21]对CE-SDS方法测定蛋白质相对分子质量和抗体纯度分析进行了联合验证实验。所有参加实验室均使用贝克曼库尔特公司的PA800仪器和CE-SDS试剂盒,多个不同实验室的不同的操作者,采用同样的分析方法对同一IgG标准品进行分析。统计结果显示,还原IgG样品的3个主要峰,重链、轻链和非糖基化重链的迁移时间RSD均小于2%,峰面积RSD均小于9%,说明该方法可满足药物质控的标准要求,可以作为抗体药物的质控方法。
为了提高CE-SDS方法的分析通量,Guttman等[22]还尝试使用多通道毛细管进行抗体药物纯度分析。其他基于毛细管电泳技术而进一步微型化的芯片电泳也已经出现[23-25]。芯片电泳具有速度更快,通量更高的特点和优势,可被用于抗体药物生产过程中的克隆筛选、细胞培养以及下游纯化过程的优化等环节。然而,由于芯片电泳的分辨率和重现性相比于常规CE-SDS方法还有一定的差距,目前还仅限于过程监控,而在单抗产品的质控时仍然需要采用CE-SDS的方法。
2 单克隆抗体药物的等电点及电荷异质性分析
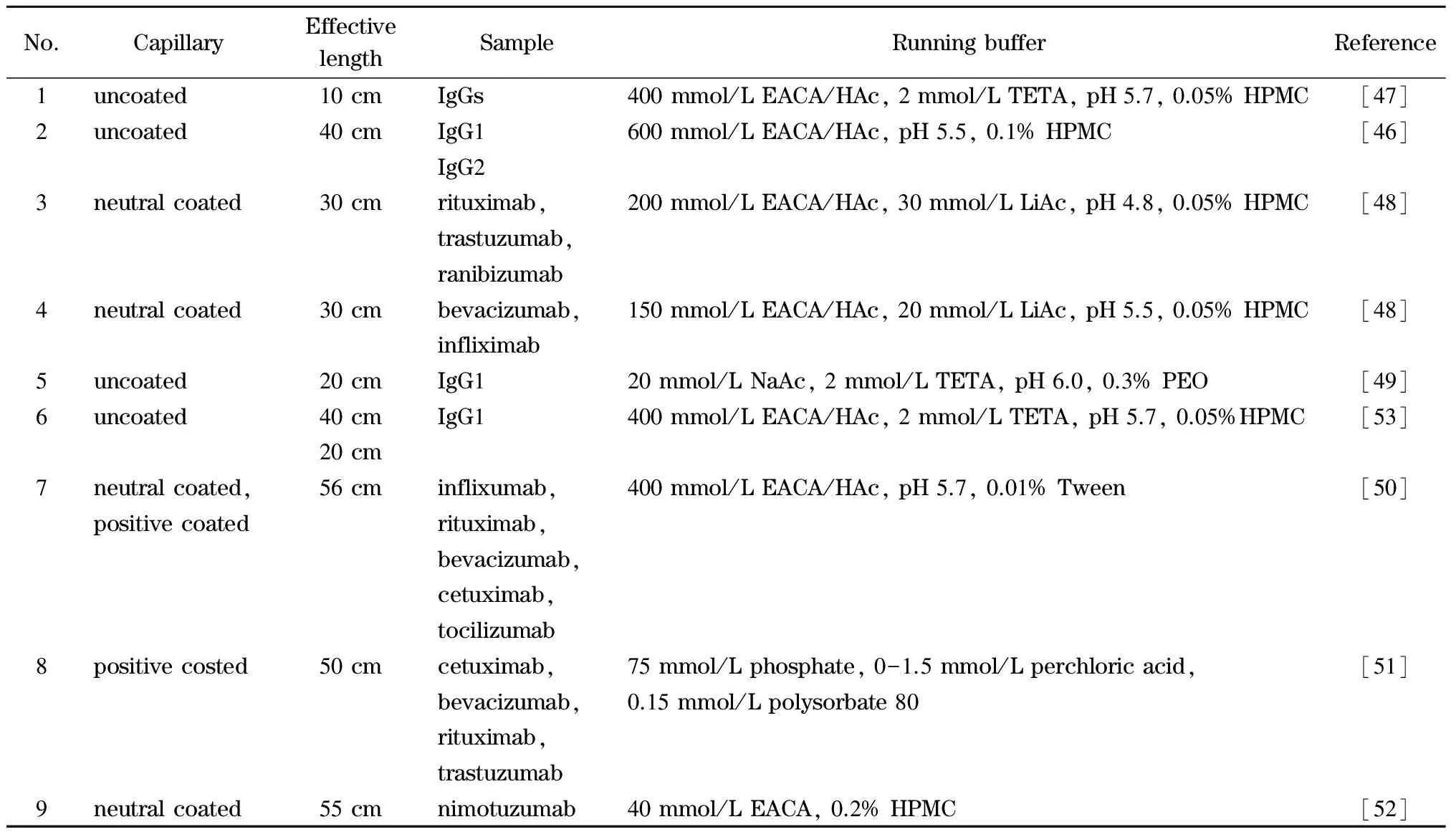
抗体的电荷异质性主要来源于氨基酸的修饰和变化,如去酰胺化产生羧基形成了抗体的酸性变异体;相反,天冬氨酸-甘氨酸容易形成琥珀酰胺而成为抗体的碱性变异体;其他修饰如谷氨酸环化形成焦谷氨酸、糖基的唾液酸化、C端赖氨酸的异质性、二硫键的氧化等都会影响抗体的电荷异质性及等电点。等电聚焦(isoelectric focusing, IEF)电泳可以根据等电点的不同分离这些蛋白质变异体,给出蛋白质电荷异质性分布的信息。IEF方法原理复杂,技术难度高,现有的IEF方法也具有与SDS-PAGE相似的局限性,如自动化程度低,重复性差,分辨率有限,线性范围窄,无法准确定量。离子交换色谱(ion exchange chromatography, IEC)是另一种蛋白质变异体分离的技术,它具有可直接收集组分,与MS结合可进行结构鉴定的优势。然而IEC方法的分辨率有限,无法保证各个变异体的高效分离。毛细管电泳技术具有分离效率高、模式多、适合蛋白质等极性物质分析的优势。目前,在生物制药领域,主要有3种基于毛细管电泳的技术用于单抗药物的等电点及电荷异质性分析,即毛细管等电聚焦法(CIEF)、全柱成像毛细管等电聚焦法(image-CIEF, iCIEF)和毛细管区带电泳法(CZE)。
2.1 毛细管等电聚焦法
CIEF是在毛细管中进行的基于等电点差异分离蛋白质及其变异体的方法。为防止蛋白质吸附和产生电渗流影响聚焦,CIEF实验需要使用中性涂层毛细管。具体过程为:将样品和两性电解质混合进样,毛细管两端分别放置在酸性和碱性溶液中。在电压作用下,两性电解质在毛细管内形成pH梯度,当样品迁移到其等电点位置时停止迁移,由此产生非常窄的样品聚集区带。因不同等电点的蛋白质将聚集在不同的位置,故形成各个组分独立的蛋白质区带而彼此分离。经过上述进样、聚焦后,再采用压力迁移或化学迁移使各组分移动到检测窗口,完成在线检测。CIEF方法具有分辨率高、自动化、样品和试剂消耗少、可准确定量等优势,是单抗以及其他蛋白质药物电荷异质性分析和等电点测定的理想方法[26-29]。
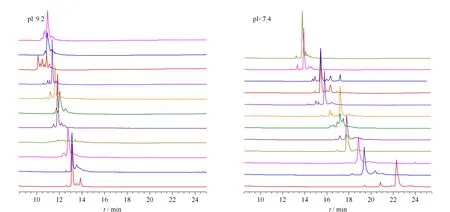
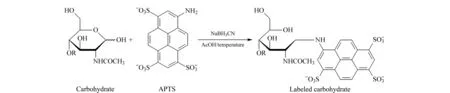
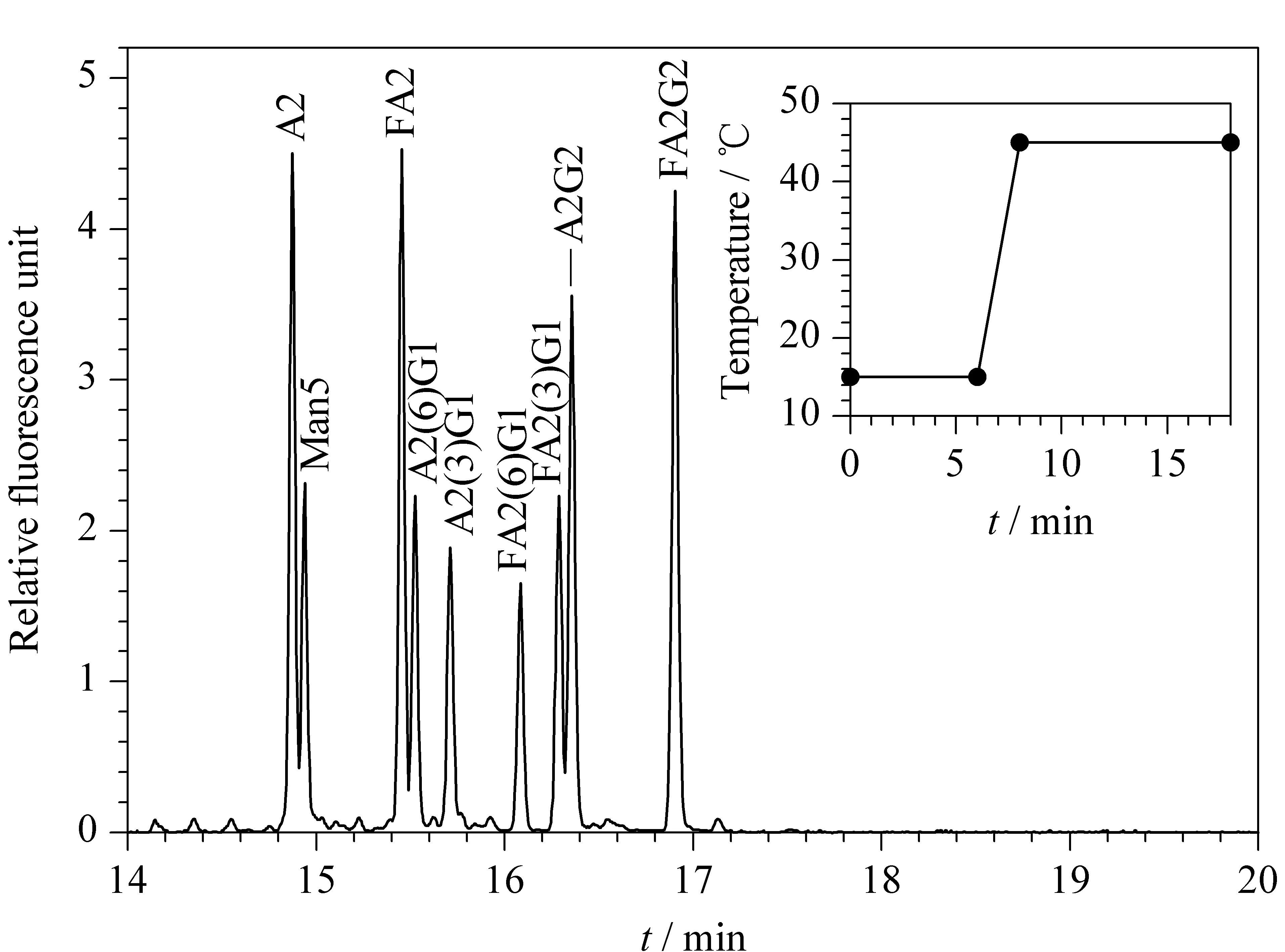
1992年,罗氏制药公司的Costello等[30]首次报道了采用CIEF技术对抗体药物的表征。CIEF技术解决了IEF中预制聚丙烯酰胺或琼脂糖凝胶在pH 7~10范围内不稳定的问题,可对抗体药物发生脱酰胺或脱糖基后产生的变异体进行准确的定量和pI测定。他们因此提出CIEF方法将会成为抗体药物表征和稳定性研究的标准方法。此后,基因泰克等公司及科研机构陆续发表了CIEF方法用于抗体药物分析的研究工作[31-35]。Lin等[34]实现了对抗体药物酸性和碱性变异体的分离及C端赖氨酸变异体和糖谱的分析。Cao等[35]对曲妥珠单抗的类似药进行电荷异质性、稳定性和批次一致性分析。雅培公司的Bonn等[36]通过使用再生冲洗液和聚乙烯醇(polyvinyl alcohol, PVA)涂层毛细管等优化的条件,使CIEF方法能够连续运行100针以上,并保持高度的重复性。Mack等[37]对CIEF方法的实验参数进行了进一步优化:如将窗口狭缝由8变成2,迁移方式由压力迁移转为化学迁移来提高分辨能力;在两性电解质溶液中加入尿素来增加蛋白质的溶解性;提高入口和出口缓冲瓶中磷酸和氢氧化钠的浓度和添加阳极稳定剂(pI <3)、阴极稳定剂(pI>10)来防止极端pI样品的损失。经过优化的CIEF方法可以在4 图 3 单克隆抗体的典型CIEF电泳图[37] Fig. 3 Typical electropherogram of mAb by CIEF[37] 2011年,美国和瑞士的10家生物制药公司的12个实验室[38]开展了对CIEF方法的国际联合验证。所有参加的实验室均使用贝克曼库尔特公司的PA800仪器、CIEF启动试剂盒和两个不同批次的两性电解质。各实验室对同一单抗样品进行分析,考察6个变异体的pI值以及含量百分比。在不同实验室、不同操作者、不同批次的两性电解质的条件下,分析结果获得高度的重复性。6个变异体pI值的RSD均小于0.5%,峰面积百分比的RSD均小于4.4%。且不同批次的两性电解质分析得到的峰面积百分比结果高度一致。验证结果支持使用CIEF方法作为生物药质控的标准方法。2013年,美国药典收录CIEF方法作为曲妥珠单抗和利妥昔单抗电荷异质性分析的方法[39]。CIEF技术不仅可分析单抗,对ADC药物进行高分辨的电荷异质性分析也已有报道[40]。 iCIEF是分离、聚焦完成后,直接采集蛋白质峰位移图像的实时检测方法。该方法无需将蛋白质组分迁移至检测窗口,且可使用短毛细管(5 cm有效长度),因此分析速度大大加快。 Zhang等[41]用iCIEF方法对来自美国、欧洲和印度市场上的利妥西单抗原研药及类似药进行分析对比,相比于美国和欧洲的原研药,来源于印度市场的类似药中,Fc/2(可结晶片段)的碱性变异体增多,轻链和Fd(Fab中抗原结合区的部分重链片段)的酸性变异体峰有缺失。基于上述结果分析得出,Fc/2中增加的碱性变异体为未切除的赖氨酸残基,而轻链和Fd中缺失的酸性峰为去酰胺化程度低造成的。基因泰克公司的Kaschak等[42]采用iCIEF法对单抗药碱性变异体进行分析,证明了培养基中铜离子与C端脯氨酸酰胺化引起的碱性变异体变化存在相关性。于传飞等[43]使用iCIEF方法对单克隆抗体进行电荷异质性分析,配合恰当的酶切处理,可判断单克隆抗体产品主要电荷异质性的来源。该方法为保证单克隆抗体药物生产工艺的稳定性及产品的质量控制提供了有效手段。近期,Goyon等[44]以美国食品药品监督管理局(FDA)和欧洲药品管理局(EMA)批准的23种单克隆抗体药物(等电点在6.1~9.4范围)作为研究对象,比较了iCIEF和阳离子交换色谱(cation-exchange chromatography, CEX)方法。结果显示iCIEF方法分析结果中酸性变异体的含量均明显高于CEX方法的分析结果。这是首次将色谱和毛细管电泳两种技术同时应用于多种单抗的电荷异质性分析。 同时参与了CIEF方法联合验证的相同的12个实验室也组织开展了对iCIEF方法的国际联合验证[45]。结果表明不同变异体的pI值的RSD均小于0.8%,峰面积百分比均小于11%。虽然iCIEF方法的重现性数据比CIEF方法略差,但仍然可以满足生物制药分析的要求,可作为CIEF方法的补充,用于单抗药物的过程分析和质量控制。 毛细管区带电泳是最简单、最常用的毛细管电泳模式。它使用自由溶液,根据分析物电荷/质量(荷质比)的差异进行分离。在单克隆抗体的电荷异质性分析中,由于抗体的变异体大多来源于氨基酸的修饰和变化,各个变异体间质量的差别不大,差异主要体现在表面电荷的不同,因此CZE分离模式的结果与基于等电点分离的等电聚焦模式具有相似性,但其较CIEF方法更简单、方便。CZE方法用于抗体药物电荷异质性分析的另一个优势在于它最容易实现与质谱检测器的连接。CZE与MS联用,可以依靠质谱对CE分离的各个电荷变异体进行结构分析,为变异体的鉴定提供重要的信息。自2010年起,CZE方法在生物制药行业备受关注,辉瑞、罗氏等众多制药公司均采用CZE方法进行抗体药物的快速电荷异质性分析。 图 4 CZE和iCIEF方法分离5种单克隆抗体的电泳图比较[46]Fig. 4 Comparison of electropherograms of five mAbs by CZE and iCIEF[46]A: acidic peak; M: main peak; B: basic peak; H: histidine in sample matrix. 辉瑞公司的He等[46,47]使用熔融石英毛细管,对CZE单抗药电荷异质性分析的电泳条件进行了考察,指出缓冲液的种类和pH是变异体分离的关键因素。他们选择pH 4.5~6.0的范围进行优化,以两性电解质ε-氨基己酸(ε-aminocaproic acid, EACA)作为缓冲液,可以降低抗体在毛细管壁的吸附。通过添加三乙烯四胺(triethylenetetramine, TETA)和羟丙基甲基纤维素(hydroxypropyl methyl cellulose, HPMC)作为动态涂层,可进一步降低管壁的吸附,改善分离效率和重现性。在400 mmol/L EACA、0.05% HPMC和适当的pH条件下,使用40 μm内径、10 cm有效长度的毛细管,对多种抗体药物均可在3~5 min内实现快速电荷异质性分析,图4为CZE和iCIEF方法分析5种单抗药物的比较。CZE方法的分离效率与iCIEF方法相当(CZE与iCIEF的峰镜像对应),而分离的时间比iCIEF方法还要快一倍[46]。样品缓冲液的选择也会对分离结果产生影响,在IgG2的分离中,向样品缓冲液中加入尿素,由于尿素打开了二硫键异构体的折叠,IgG2主峰出现分叉,二硫键异构体得以分离[47]。目前报道的针对各种单抗药物的CZE电荷异质性分析方法中,大多基于He等报道的上述方法,采用pH 4.5~6.0范围的EACA作为缓冲液这一基本条件。不同方法间的差别主要来自动态涂层试剂和添加剂(如TETA、HPMC及吐温等)的选择和浓度的不同。由于缓冲液EACA的紫外吸收低,允许使用214 nm波长检测,相比使用280 nm检测的CIEF和iCIEF方法,灵敏度有所提高。 Ed Garza等[48]基于He的上述工作建立了CZE方法平台,选择不同的缓冲体系对多种抗体药物及其类似药进行分析,并将此CZE平台方法用于抗体药物批放行及产品研发中。他们使用中性涂层毛细管,降低了抗体在毛细管壁的吸附。使用醋酸调节pH值、醋酸锂平衡缓冲容量,增加电导率和离子强度、改善分离。最终确定利妥西单抗(rituximab)、曲妥珠单抗(trastuzumab)、雷珠单抗(ranibizumab)及其类似药分离的最佳缓冲条件为200 mmol/L EACA/HAc, 30 mmol/L LiAc, pH 4.8, 0.05% HPMC;贝伐单抗(bevacizumab)、英夫利昔单抗(infliximab)及其类似药分离的最佳缓冲条件为100 mmol/L EACA/HAc, 20 mmol/L LiAc, pH 5.5, 0.05% HPMC。方法稳定性好,在优化的条件下,一根中性涂层毛细管可以提供120~150次稳定的分析结果。然而,如需获得各变异体的结构信息以评价其生物相似性,仍需要采用其他技术的辅助。 Lin实验室[49]报道了另外一种CZE缓冲液体系,20 mmol/L NaAc, 2 mmol/L TETA, pH 6.0, 0.3% PEO,该方法对IgG碱性变异体的分离优于He的方法,可以分离出两个碱性变异体。他还指出CZE方法除了可区分碱性端赖氨酸变异体,还可以快速分离Fab和Fc、完整抗体及相关物质。在其他相似的研究工作中,除了使用熔融石英毛细管和中性涂层毛细管,也有使用阳离子涂层毛细管进行抗体药物的CZE电荷异质性研究的报道[50,51]。另外,由于CZE方法具有简单快速、分辨率高的优势,故CZE方法也被用于抗体药物的鉴别。郭玮等[52]利用CZE方法对尼妥珠单抗进行了专属性鉴别分析,证明该方法对于尼妥珠以及其他8种单克隆抗体的特异性鉴别有效。目前,尚无CZE电荷异质性分析的商品化试剂盒,我们对已报道的CZE方法的主要分离条件进行了总结(见表3)。 表 3 CZE电荷异质性分析的分离条件Table 3 CZE conditions for mAb charge variant analysis 与CIEF和iCIEF方法的多企业联合验证目的相似,为了评估CZE在生物制药行业应用的可行性,罗氏制药的Moritz等[53]组织来自美国、瑞士、德国的11家实验室开展了CZE方法的联合验证。所有参加实验室均使用贝克曼库尔特公司的PA800 Plus或Enhanced仪器。他们先采用同一CZE条件对pH 7.4~9.5的23种单抗进行分析,结果表明该方法对多种单抗药物具有普适性,结果见图5。他们还对50 cm×50 μm和40 cm×30 μm两种长度/内径毛细管的分离条件进行了比较。结果显示较长的毛细管对杂质分离的分辨率比短毛细管略好,而短毛细管则可以提供更好的稳定性、更快的速度(3.5~7.5 min)和高通量分析的能力。在联合验证实验中,选用了样品中酸性最强(pI最小)、分离时间最长的单抗作为样品。如图6所示,将CZE方法的分析结果与iCIEF和IEC方法进行了对比,3种方法主峰区的CPA%(校准峰面积百分比)结果相似,分别为61.5%(11.1+28.6+21.8=61.5)、61.6%和64.7%。但CZE方法的分辨率最高,主峰区可分离出3个峰(CZE与iCIEF/IEC方法的峰镜像对应)。特别指出的是,所有参加验证的操作人员在此次验证前均未接受额外的培训,但却获得了非常好的重复性结果,RSD为1%左右。该验证结果充分证明了CZE方法作为一种抗体药物电荷异质性分析平台方法的可行性。 图 5 同一CZE方法对23种单抗药物的电荷异质性分析[53]Fig. 5 Charge variant analysis of 23 mAbs by the same CZE condition[53] 图 6 CZE、iCIEF和IEC方法分析同一单抗的电荷异质性[53]Fig. 6 Charge variant analysis of the same mAb by CZE, iCIEF and IEC[53] 尽管上述联合验证证实了CZE方法简单可靠,然而使用同一个分离条件很难保证对所有的抗体药物都达到最佳的分离效果。因此,Moritz等[54]又继续展开了针对大量实验参数的优化研究,他们依据DoE(Design of Experiment)原则优化参数,选用分辨率、峰宽、峰个数等多项标准评判参数优化的结果,提高了13种抗体及相关重组蛋白质的CZE分离效果。经过大量的研究得出结论,在CZE对抗体及其相关形式药物(如双特异性抗体和其他融合蛋白质)的电荷异质性分析中,TETA的浓度、pH值、聚合物添加剂(HPMC或羟丙基纤维素(HPC))和其他添加剂(乙腈或丁醇胺)这几个参数为需要优化的关键参数。带正电荷的TETA与毛细管壁的负电荷相互作用,通过降低电渗流及分析物在管壁的吸附来提高分辨率。pH值通过影响电荷/质量比而直接影响分析物的迁移速度,对分离结果影响极大。HPMC的黏度大于HPC,会延长分离时间。作为溶剂的乙腈,当其含量适量时,可通过降低分析物之间的疏水相互作用而提高分离度;而当乙腈含量过高时,会使溶液的电导率和组分的溶解性改变而降低分离度。带电添加剂(如丁醇胺)则通过降低电渗流、分析物与管壁的相互作用(改变分析物的电荷/质量)提高分离度。Moritz等的工作为不同样品的CZE方法优化提供了重要指导,也使得CZE电荷异质性分析方法的应用范围由抗体药物拓展到其他抗体相关的重组蛋白质。 单克隆抗体的重链Fc端在Asn297位置有一个糖基化位点,细胞培养过程中,会产生多种寡糖变异体。由于寡糖对药代动力学、生物活性、稳定性和免疫原型具有重要影响,法规机构将抗体中的寡糖视为关键质量属性(critical quality attribute, CQA)。如核心岩藻糖缺失和双分支N-乙酰氨基葡萄糖(N-acetylglucosamine, GlcNAc)的存在会增加ADCC活性,降低唾液酸含量也会引起ADCC活性增加。双天线糖链的末端半乳糖残基会影响补体依赖的细胞毒性(complement-dependent cytotoxicity, CDC)等。抗体药物中寡糖的异质性来源于表达体系、克隆、培养基、温度和时间等生长条件。因此,在生产过程和批放行中对寡糖进行定量和监控非常重要。单克隆抗体中寡糖谱图分析的主要策略有:(1)对还原或非还原完整抗体自上而下的表征;(2)糖肽水平的表征;(3)对酶解释放的寡糖链的表征。但是由于其他氨基酸修饰、氧化等异质性的干扰,寡糖异质性分析多采用把寡糖链酶解下来分析的方法。可用于寡糖分析的方法有NMR、MS、HPLC和CE,其中比较常用的是高效阴离子交换色谱-脉冲安培检测法(HPAEC-PAD)[55,56]、HPLC-荧光检测法[57-59]和毛细管电泳-激光诱导荧光检测法(CE-LIF)[60-65]。这些方法均可以提供高分辨和高灵敏的分析。离子交换色谱方法的优点在于无需衍生化,但其在特异性、定量和多功能性方面有一定局限。HPLC和CE技术则均需要对酶解释放的寡糖进行衍生化。CE可以对寡糖的位置异构体和连接异构体进行高效分离,且在分析速度上比HPLC更具有优势[66]。生物制药工业中对于产品质量属性的评估通常会采取互相补充的多种技术,来避免单一技术检测带来的风险。 图 7 APTS与寡糖的反应Fig. 7 Reaction between 8-aminopyrene-1,3,6-trisulfonic acid, trisodium salt (APTS) and oligosaccharide CE方法糖基分析的标记试剂需要满足以下两个条件:1.带有一个或多个电荷,使得不易带电的寡糖分子带上电荷,有利于CE的分离;2.具有荧光发色基团,使标记后的寡糖可以在指定激发波长下具有荧光信号,提供高灵敏度的检测。可用于CE方法的荧光标记试剂有8-氨基芘-1,3,6-三磺酸三钠盐(APTS)、8-氨基萘磺酸-1,3,6-三磺酸三钠盐(ANTS)和2-氨基苯甲酸(2-AA)等。2-AA所带负电荷少、出峰时间慢,可以分离低丰度的唾液酸化和高甘露糖化的寡糖[67]。但因没有对应波长的商品化激光器而使其应用受限。采用APTS标记糖时分离速度快、理论塔板数高,具有商品化的激光诱导荧光光源和检测器(488 nm激发/520 nm发射),故APTS是单克隆抗体药物寡糖分析最常用的标记试剂。APTS结构中的芘结构提供了荧光响应,3个磺酸基阴离子提高了CE分离的效率和速度,氨基与寡糖链末端的缩醛基在还原剂的作用下反应完成标记。标记反应过程见图7。由于每个寡糖链只有末端的一个缩醛基,可以保证每个寡糖上只标记一个APTS,因此可根据荧光信号对寡糖进行定量分析,APTS标记的寡糖分析灵敏度可达0.4 nmol/L[62]。 1999年,基因泰克公司的Nashabeh和Ma[68]采用PNGase F酶酶解释放寡糖链,再使用APTS标记后,对几种主要的N-寡糖G0、G1、G1′和G2(结构见图8)进行了CE-LIF分离检测,以验证CE-LIF方法作为利妥昔单抗药常规放行检测的可行性。结果表明,CE-LIF方法不仅可以实现不同链长的寡糖的分离,还可以分离G1和G1′两种糖的位置异构体。其中位置异构体的鉴定是通过β-N-乙酰氨基己糖苷酶和α-1-2,3甘露糖苷酶逐级酶解进行验证的。 图 9 糖蛋白测试品中的N-寡糖经APTS标记后的CE-LIF分析电泳图[70]Fig. 9 CE-LIF analysis trace of the APTS-labeled N-glycan profile of the protein test article[70] 继贝克曼库尔特公司推出基于APTS标记的CE-LIF糖分析试剂盒后,大部分生物制药公司均采用商品化试剂盒进行抗体药物中寡糖的检测[69]。2013年,安进公司[70]组织了来自美国、欧洲和中国的20个生物制药企业、大学及国家法规实验室开展了抗体药物中寡糖分析的多实验室联合验证。所有参加实验室均使用贝克曼库尔特公司的PA800 Plus仪器和糖分析试剂盒。结果表明该方法的重现性好,显示了在生物制药工业的克隆筛选、过程开发和批放行中建立寡糖分析平台的可行性。联合验证采用了预标记的同源寡糖梯度标准品、预标记的高甘露糖化的寡糖混合物、非岩藻糖化的寡糖混合物、岩藻糖化的寡糖混合物、预标记的寡糖测试品和糖蛋白测试品6个样品。考察了在不同实验室、仪器、操作者和日期下,方法的重复性和重现性。经过酶切和标记等前处理后,糖蛋白测试品中N-寡糖的CE分离结果见图9。根据计算的糖单位(glucose unit, GU)值使用N-寡糖数据库进行峰的指认。对丰度大于0.1%的20个糖型峰的重复性(实验室内)和重现性(实验室间)进行计算,其平均面积重复性为0.95%、重现性为4.60%。平均迁移时间重复性为0.06%,重现性为2.24%。结果表明,CE-LIF方法可以提供高通量和高灵敏度的寡糖鉴定和相对定量。但方法的不足是会有共洗脱的糖型,比如FA2和A2(6)G1同时出峰。 为解决部分寡糖共洗脱的问题,Guttman等[71]考察了毛细管温度对各种寡糖分离的影响,发现对于不同寡糖,获得最佳分离度所需的温度可能不相同。因此为了增加CE-LIF方法寡糖分离的选择性,他们建立了梯度升温的分离方法。以生物制药中很受关注的岩藻糖化寡糖、非岩藻糖化寡糖和高甘露糖化寡糖的混合物为样品,进行了温度梯度由15 ℃到45 ℃条件下的分离。结果显示,通过温度变化可以实现几对共洗脱的寡糖的分离(如FA2和A2(6)G1,A2G2和FA2(3)G1)(见图10)。 图 10 梯度升温CE方法对APTS标记的生物药寡糖的分离[71]Fig. 10 Separation of APTS labeled glycans of bio-therapeutics interest by temperature gradient capillary electrophoresis[71] 2011年,瑞士药监局[72]使用CE-LIF方法对瑞士市场上的16种单克隆抗体药物中的寡糖进行监管检测。结果显示,同一厂家的药物的批次一致性很好,说明厂家都对产品进行了严格的质控,但是不同厂家的药物的寡糖差别较大,这些差别主要来源于细胞培养条件的影响。因此,在培养基的优化、克隆筛选等生产过程中对寡糖的检测越来越受到重视,这也意味着对高通量快速寡糖分析方法的需求日益增加。为此,Guttman等[73]发明了利用磁珠对糖分析样品进行前处理的方法。使用磁珠进行酶切和标记,免除了所有前处理过程中的离心以及真空离心的操作,节省了大量的时间。文中以单克隆抗体等3种糖蛋白为模型,证明了该快速方法与传统前处理方法在寡糖释放和标记效率上具有相似的效率。方法具有高度的重复性,并且非常容易实现自动化分析。随后Guttman等[74]在另一篇文章中报道了在自动化工作站中进行96个样品的全自动前处理和CE分离的工作,并优化了CE分离的条件,使分离时间进一步缩短至3 min。基于Guttman的上述工作,2017年Sciex公司(原贝克曼库尔特公司)推出了快速糖分析试剂盒,该试剂盒采用磁珠辅助的前处理流程和快速分离的缓冲体系。酶切、标记和清洗等所有前处理过程所需的时间缩短至1 h(前处理流程见图11),采用经过优化的缓冲体系,使CE分离的时间缩短至5 min以内。 图 11 磁珠辅助法进行N-寡糖分析的样品前处理流程图[73]Fig. 11 Full magnetic bead based sample preparation workflow for N-glycosylation analysis[73] 此外,基于毛细管电泳原理进行寡糖分离的芯片电泳也被用于抗体药物的克隆筛选中,可提供快速(约1 min)和高通量的寡糖分析。但其样品前处理仍需要3~4 h,且芯片技术的分辨率和重复性仍然低于常规毛细管电泳技术。 在CE-LIF方法中,通常根据荧光检测得到的电泳峰的校准峰面积对各种寡糖的相对含量进行定量分析。若需对糖型进行鉴定,则需要借助其他手段,如(1)使用寡糖标准品鉴定;(2)根据寡糖梯度标准品计算待测寡糖的GU值来鉴定;(3)使用外切酶逐级酶解寡糖进行鉴定[68]; (4)使用制备型HPLC收集,再将收集得到的组分经MS鉴定后,作为CE的标准品进行鉴定[75]; (5)CE-MS联用技术直接对CE分离得到的寡糖进行质谱鉴定[76]。其中计算GU值的鉴定方法最为简单快速。最近,Guttman课题组[77]设计了基于3种内标糖计算GU值的方法。选用麦芽2糖、麦芽3糖和麦芽15糖3种糖作为内标物与被分析样品混合后同时进样,根据3个内标糖的迁移时间建立虚拟的寡糖梯度,用于计算待测寡糖的GU值。特别值得指出的是,这样一套基于3个内标的GU值计算方法并不受毛细管长度、分离电压以及进样量等实验条件变化的影响,避免使用寡糖梯度标准品单独进样,也节省了分析时间。在Sciex公司的快速糖分析试剂盒中,使用了该快速GU值鉴定的方法。 在单克隆抗体药物的分析和质控中,毛细管电泳技术已经成为不可或缺的必要手段。利用毛细管凝胶电泳进行纯度(或大小异质性)分析和N-寡糖分析,利用毛细管等电聚焦/全柱成像毛细管等电聚焦/毛细管区带电泳进行等电点及电荷异质性分析,这些方法已经广泛用于单克隆抗体的产品质控、过程开发及批放行中。随着越来越多种类的蛋白质药物的出现,相关分析的技术开发热点将集中在以下几个方面:(1)快速、高通量分析技术的开发,以满足大量样品的检测和克隆筛选等生产环节的需求。(2)建立针对其他蛋白质药物(抗体片段、双特异性抗体、抗体聚合体、抗体偶联药物及其他融合蛋白质)的分析方法。例如开发适合相对分子质量更小的或更大的蛋白质药物分析的凝胶缓冲液、适合高糖基化蛋白质药物分析的凝胶缓冲液、适合极度酸性蛋白质和高度修饰蛋白质分析的CIEF方法等。(3)CE-SDS/CIEF/CZE与MS技术离线或在线联用方法的开发,以解决经电泳分离得到的杂质、电荷变异体和寡糖的鉴定问题。其中CE-MS联用技术对单抗药物的肽谱序列、翻译后修饰、糖肽和完整蛋白质电荷异质性等多水平表征的应用已有报道,我们将另外对这部分进展进行综述。 致谢感谢丽珠医药的邓钦培博士在文章撰写过程中的建议和指导。 参考文献: [1]Mullard A. J Nat Rev Drug Discov, 2012, 11(6): 426 [2]Guo W, Wang L, Gao K. Chinese Journal of New Drugs, 2014, 23(20): 2351 郭玮, 王兰, 高凯. 中国新药杂志, 2014, 23(20): 2351 [3]Wang L, Zhu L, Xu G L, et al. Chinese Pharmaceutical Journal, 2014, 49(23): 2058 王兰, 朱磊, 徐刚领, 等. 中国药学杂志, 2014, 49(23): 2058 [4]Gahoual R, Beck A, Leize-Wagner E, et al. J Chromatogr B, 2016, 1032: 61 [5]Cao W R, Zhang S G, Wang X J, et al. Chemistry & Bioengineering, 2012, 29(6): 20 曹卫荣, 张世光, 王晓婧, 等. 化学与生物工程, 2012, 29(6): 20 [6]Cohen A S, Paulus A, Karger B L. Chromatographia, 1987, 24: 15 [7]Guttman A. Electrophoresis, 1996, 17: 1333 [8]Pharmacopoeia Commission of the People’s Republic of China. Pharmacopoeia of the People’s Republic of China, Part 3. Beijing: China Medical Science Press, 2015: 42 国家药典委员会. 中华人民共和国药典, 三部. 北京: 中国医药科技出版社, 2015: 42 [9]Shi Y, Li Z, Lin J. Anal Methods, 2012, 4: 1637 [10]Zhang J, Burman S, Gunturi S, et al. J Pharm Biomed Anal, 2010, 53: 1236 [11]Rustandi R R, Washabaugh M W, Wang Y. Electrophoresis, 2008, 29: 3612 [12]Li W, Yang B, Zhou D, et al. J Chromatogr B, 2017, 1048: 121 [13]Wang W B, Wang L, Yu C F, et al. Chinese Pharmaceutical Journal, 2016, 51(1): 46 王文波, 王兰, 于传飞, 等. 中国药学杂志, 2016, 51(1): 46 [14]Li M, Zhu L, Wu G, et al. Chinese Pharmaceutical Journal, 2016, 51(13): 1091 李萌, 朱磊, 武刚, 等. 中国药学杂志, 2016, 51(13): 1091 [15]Ahuja S, Jimidar M I. Capillary Electrophoresis Methods for Pharmaceutical Analysis. 1st ed. Oxford: Elsevier Inc, 2008: 369 [16]Hunt G, Nashabeh W. Anal Chem, 1999, 71: 2390 [17]Salas-Solano O, Tomlinson B, Du S, et al. Anal Chem, 2006, 78: 6583 [18]Michels D A, Brady L J, Guo A, et al. Anal Chem, 2007, 79: 5963 [19]Michels D A, Parker M, Salas-Solano O. Electrophoresis, 2012, 33: 815 [20]Han M, Phan D, Nightlinger N, et al. Chromatographia, 2006, 64: 335 [21]Nunnally B, Park S, Patel K, et al. Chromatographia, 2006, 64: 359 [22]Szekrényes A, Roth U, Kerékgyártó M, et al. Anal Bioanal Chem, 2012, 404: 1485 [23]Legmann R, Schreyer H B, Combs R G, et al. Biotechnol Bioeng, 2009, 104: 1107 [24]Chen X, Tang K, Lee M, et al. Electrophoresis, 2008, 29: 4993 [25]Antes B, Oberkleiner P, Nechansky A, et al. J Pharm Biomed Anal, 2010, 51: 743 [26]Silverman C, Komar M, Shields K, et al. J Liq Chromatogr, 1992, 15(2): 207 [27]Schwer C. Electrophoresis, 1995, 16: 2121 [28]Liu X, Sosic Z, Krull I S. J Chromatogr A, 1996, 735: 165 [29]Kilar F. Electrophoresis, 2003, 24: 3908 [30]Costello M A, Woititz C, DeFeo J, et al. J Liq Chromatogr, 1992, 15: 1081 [31]Hunt G, Moorhouse K G, Chen A B. J Chromatogr A, 1996, 744: 295 [32]Hunt G, Hotaling T, Chen A B. J Chromatogr A, 1998, 800: 355 [33]Suba D, Urbanyi Z, Salgo A. J Pharm Biomed Anal, 2015, 114: 53 [34]Lin J, Tan Q, Wang S. J Sep Sci, 2011, 34: 1696 [35]Cao J Z, Sun W, Gong FF, et al. Electrophoresis, 2014, 35: 1461 [36]Bonn R, Rampal S, Rae T, et al. Electrophoresis, 2013, 34: 825 [37]Mack S, Cruzado-Park I, Chapman J. Electrophoresis, 2009, 30: 4049 [38]Salas-Solano O, Babu K, Park S, et al. Chromatographia, 2011, 73: 1137 [39]The United States Pharmacopieial Convention. United States Pharmacopeia. New York: United States Pharmacopeia Press, 2013: 37 [40]Maedaa E, Urakamia K, Shimurab K, et al. J Chromatogr A, 2010, 1217: 7164 [41]Zhang Z, Perrault R, Zhao Y, et al. J Chromatogr B, 2016, 1020: 148 [42]Kaschak T, Boyd D, Lu F, et al. mAbs, 2011, 3(6): 577 [43]Yu C F, Guo W, Wang L, et al. Chinese Journal of Pharmaceutical Analysis, 2014, 34(7): 1212 于传飞, 郭玮, 王兰, 等. 药物分析杂志, 2014, 34(7): 1212 [44]Goyon A, Excoffier M, Janin-Bussat M-C, et al. J Chromatogr B, 2017, 1065/1066: 119 [45]Salas-Solano O, Kennel B, Park S, et al. J Sep Sci, 2012, 35: 3124 [46]He Y, Isele C, Hou W, et al. J Sep Sci, 2011, 34: 548 [47]He Y, Lacher N A, Hou W, et al. Anal Chem, 2010, 82: 3222 [48]Espinosa-de la Garza C E, Perdomo-Abundez F C, Padilla-Calderon J, et al. Electrophoresis, 2013, 34: 1133 [49]Shi Y, Li Z, Qiao Y B, et al. J Chromatogr B, 2012, 906: 63 [50]Gassner A L, Rudaz S, Schappler J. Electrophoresis, 2013, 34: 2718 [51]Jaccoulet E, Smadja C, Prognon P, et al. Electrophoresis, 2015, 36: 2050 [52]Guo W, Wang L, Wang W B, et al. Chinese Journal of New Drugs, 2014, 23(20): 2366 郭玮, 王兰, 王文波, 等. 中国新药杂志, 2014, 23(20): 2366 [53]Moritz B, Schnaible V, Kiessig S, et al. J Chromatogr B, 2015, 983/984: 101 [54]Moritz B, Locatelli V, Niess M, et al. Electrophoresis, 2017, 38: 3136 [55]Hardy M R, Townsend R R, Lee Y C. Methods Enzymol, 1989, 179: 65 [56]Spellman M. Anal Chem, 1990, 62: 1714 [57]Nakano M, Kakehi K, Tsai M H, et al. Glycobiology, 2004, 14: 431 [58]Kamoda S, Nakano M, Ishikawa R, et al. J Proteome Res, 2005, 4: 146 [59]Naka R, Kamoda S, CIshizuka A, et al. J Proteome Res, 2006, 5: 88 [60]Kamoda S, Nomura C, Kinoshita M, et al. J Chromatogr A, 2004, 1050: 211 [61]Klockow A, Amado R, Widmer H M, et al. J Chromatogr A, 1995, 716: 241 [62]Evangelista R A, Liu M-S, Chen F-T A. Anal Chem, 1995, 67: 2239 [63]Chen F-T A, Evangelista R A. Anal Biochem, 1995, 230: 273 [64]Kamoda S, Ishikawa R, Kakehi K. J Chromatogr A, 2006, 1133: 332 [65]Szabo Z, Guttman A, Bones J, et al. Anal Chem, 2011, 83: 5329 [66]Kamoda S, Kakehi K. Electrophoresis, 2008, 29: 3595 [67]Kamoda S, Ishikawa R, Kakehi K. J Chromatogr A, 2006, 1133: 332 [68]Ma S, Nashabeh W. Anal Chem, 1999, 71: 5185 [69]Wang W B, Wang L, Wang X, et al. Chinese Journal of New Drugs, 2015, 24(20): 2312 王文波, 王兰, 王馨, 等. 中国新药杂志, 2015, 24(20): 2312 [70]Szekrenyes A, Park S, Santos M, et al. mAbs, 2016, 8(1): 56 [71]Szigeti M, Guttman A. Anal Chem, 2017, 89(4): 2201 [72]Wacker C, Berger C N, Girard P, et al. Eur J Pharm Biopharm, 2011, 79: 503 [73]Varadi C, Lew C, Guttman A. Anal Chem, 2014, 86: 5682 [74]Szigeti1 M, Lew C, Roby K, et al. J Lab Autom, 2016, 21(2): 281 [75]Hamm M, Wang Y, Rustandi R R. Pharmaceuticals, 2013, 6: 393 [76]Bunz S-C, Rapp E, Neususs C. Anal Chem, 2013, 85: 10218 [77]Jarvas G, Szigeti M, Chapman J, et al. Anal Chem, 2016, 88: 11364
2.2 全柱成像毛细管等电聚焦法
2.3 毛细管区带电泳法


3 单克隆抗体药物的N-寡糖分析


4 总结与展望
