基于氨基与表面乙烯砜基反应动力学调控配基表面密度
2018-03-29程昉李明洋何炜王汉奇1大连理工大学精细化工国家重点实验室辽宁大连116023
程昉,李明洋,何炜,王汉奇1大连理工大学精细化工国家重点实验室,辽宁 大连 116023
2大连理工大学制药科学与技术学院,辽宁 大连 116023
3大连理工大学化工学院,辽宁 大连 116023
1 引言
自组装单分子膜用于表面修饰,能够在表面引入各种功能性分子,所以被广泛应用于生物传感器1,2、酶固定化3、受体-配体相互作用4、蛋白纯化5、生物医学工程6,7等研究工作中。在以上的研究工作中,通常需要对表面生物配基密度进行精准控制,例如在蛋白纯化配基密度优化工作中,需要制备不同配基密度的表面从而对最优配基密度进行筛选5;在功能性材料的研究中,也需要对配基表面密度进行优化从而降低非特异性吸附7–9。综上所述如何实现配基表面密度控制成为了一项具有重要意义的研究工作。
目前应用最为广泛的表面密度控制方法是混合自组装,通过调控功能分子与非功能分子的浓度比对表面密度进行调控10–13。例如,Bohmler等14利用混合自组装的方法得到了一系列密度梯度的Br/CH3和NH2/CH3表面并进行了细菌粘附实验。Tomohiro等15利用混合自组装的方法分别在表面修饰了含有 1–3个 6-磺基-N-乙酰基-葡萄糖胺基团的分子并进行了多价态相互作用的研究。Liu等16通过优化混合自组装比例对固定于表面的蛋白质活性进行优化。本组之前工作17利用混合自组装对表面糖基功能分子密度进行调控,研究了葡萄糖与Con A蛋白的相互作用。然而,混合自组装的方法在应用中具有局限性,当进行混合自组装的两种分子极性相差较大时会产生相分离从而影响自组装效果,并且对于不同的自组装体系不具有普适性。
近年的研究表明利用反应进程控制的方法能够很好地控制表面密度18,19。反应进程控制法结合表面反应动力学,通过控制配体与表面基团的反应进程实现密度控制。Zhang等19利用Azide-alkyne点击化学反应实现了表面叠氮基团的密度控制。与混合自组装的方法相比,操作上更为简便,可以根据配基的不同调整反应类型,并且对于不同的体系具有普适性。
本组之前工作合成了一种VS (Vinyl Sulfone,乙烯砜基)末端的二硫化合物,利用该化合物构建了VS自组装表面,并对表面VS基团与巯基、氨基以及羟基在不同 pH条件下的反应动力学进行了研究20。该工作为通过表面反应实现表面密度控制打下了基础。我们利用自组装的方法在表面引入VS基团,以ab-NTA为生物配基模型,通过表面催化反应实现NTA密度控制。首先我们利用接触角的方法对催化 VS与氨基反应的催化剂进行了筛选,并对反应动力学进行了表征。利用反应动力学结果,得到不同配基密度的表面。最后我们利用SPR的方法对不同NTA密度表面与组氨酸标签蛋白的吸附/解离动态过程进行了研究。
2 实验部分
2.1 试剂与仪器
Nα,Nα-二(羧甲基)-L-赖氨酸(ab-NTA,97%)购买于Sigma-Aldrich;吡啶二甲酸(97.5%),1-甲基咪唑(99%)购买于 J&K(北京);三苯基膦(分析纯)购买于天津市科密欧化学试剂有限公司;HS-C10-COONHS (90%)购买于 ProChimia Surfaces;4-二甲氨基吡啶(DMAP,99%),氯化镍(分析纯),磷酸二氢钠(分析纯),乙二胺四乙酸(EDTA,分析纯)购买于阿拉丁生化科技有限公司(上海);咪唑(99%)、链霉亲和素(SA-6His)购买于索莱宝科技有限公司(北京)。其余有机溶剂均为分析纯,购买于阿拉丁生物科技有限公司。
水接触角测量使用的是接触角测量仪JC2000D1(中国,上海);表面膜电位表征使用的是NanoBrook Omni (Brookhaven Instruments,美国);表面光电子能谱(XPS)表征使用的是PHI Quantera II Scanning XPS Microprobe (清华大学摩擦学国家重点实验室提供);SPR实验均使用的是一台具有发光二极管光源(λ = 670 nm),高折射率棱镜(n = 1.61)和双通道流通单元的Biosuplar-400T SPR光谱仪(Analytical μ-Systems,德国);紫外分光光度计使用的是BioSpectrometer(Eppendorf,德国)。
2.2 自组装表面制备
2.2.1 金衬底制备
硅片在食人鱼溶液(97%浓硫酸和30%过氧化氢比例3 : 1 (V : V)。注意:食人鱼溶液对有机物有强烈腐蚀性。)中洗涤30 min后使用大量水和乙醇进行冲洗并用氮气吹干。利用 Turbo Sputter Coater K575XD (Kent,U.K.)将钛(5 nm)和金(45 nm)依次蒸镀到干净的硅片上。
2.2.2 基于VS的ab-NTA自组装表面制备
金衬底利用紫外臭氧仪清洗30 min并用乙醇冲洗。随后,配置 1.0 mmol·L−1的 1,2-二(11-(乙烯砜基)十一烷基)二硫烷溶液(二氯甲烷/乙醇 1 : 1(V : V)),其合成方法之前已有报道20。将清洗干净的衬底浸入溶液中25 °C放置12 h之后用乙醇冲洗,氮气吹干。
将 VS自组装样品浸入 ab-NTA溶液(10 mmol·L−1ab-NTA、催化剂和 10 mmol·L−1三乙胺溶于 DMSO)中进行反应。之后将样品浸入EG3-NH2水溶液(10 mmol·L−1,pH 9.5)中反应 12 h进行封闭。最后,用水和乙醇将自组装样品冲洗干净,氮气吹干,4 °C避光保存。
2.2.3 基于NHS的NTA自组装表面制备
将清洗干净的金衬底浸入 1.0 mmol·L−1的HS-C10-COONHS乙醇溶液中25 °C放置12 h,之后用乙醇冲洗,氮气吹干。NHS自组装样品浸入ab-NTA 溶液(10 mmol·L−1,pH 8.5 磷酸盐缓冲液(PBS))中反应 12 h。最后自组装样品用水冲洗干净,氮气吹干,4 °C避光保存。
2.3 SPR光谱学
为了测量SA-6His蛋白的特异性吸附量,实验步骤如下:首先通入10 min 0.1 mol·L−1NiCl2溶液进行螯合,其次通入10 min PBS缓冲液(20 mmol·L−1,pH 7.4)进行洗涤,接下来通入30 min的蛋白溶液,随后通入10 min 含0.5 mol·L−1咪唑的PBS溶液进行洗脱,再10 min PBS进行洗涤,之后通入5 min含20 mmol·L−1EDTA的PBS溶液对表面进行再生,最后同入10 min PBS溶液对表面进行洗涤。在测量BSA非特异性吸附时,洗脱溶液更换为0.01 mol·L−1NaOH溶液。
2.4 吸附平衡和热力学
含有不同蛋白质浓度(0.001–0.5 mg·mL−1)的缓冲液通过传感器表面30 min。达到吸附平衡后表面进行再生,利用紫外分光光度计检测280 nm处吸光度确定蛋白质浓度。等温吸附曲线利用Langmuir方程进行拟合:

其中Req和Ceq分别表示平衡吸附时的SPR响应值和平衡蛋白浓度。Rmax表示最大结合量,Kd表示解离常数。
3 结果与讨论
3.1 催化剂和催化条件筛选
以ab-NTA为生物活性配基,我们对表面VS基团与ab-NTA反应进行了催化剂筛选。由于氨基与乙烯砜基的反应属于氮参与的迈克尔加成反应21,本文选择了五种常用迈克尔加成反应催化剂22(如图1所示),并采用静态水接触角对其催化性能进行了表征。VS表面疏水性较强,随着反应的进行表面逐渐被 ab-NTA的羧基覆盖,疏水性逐渐降低,表面静态水接触角逐渐降低。
不同催化剂催化2和6 h后的VS表面静态水接触角变化值如图 2所示。由结果可以看出,除三乙胺之外的其余四种催化剂均具有催化该反应的能力,其中吡啶二甲酸的催化效率最高,反应6 h后接触角降低17.0°,添加三乙胺基本没有反应,表明在反应体系中添加的三乙胺不会对催化结果产生影响。随后对吡啶二甲酸的用量进行了优化,在0.1%–20%范围内选择了5个浓度进行了试验,催化2 h后检测接触角变化值,结果如图2b所示,结果显示催化剂用量在 10%之前催化效率随催化剂用量增加而逐渐升高,当催化剂用量大于 10%时,催化效率基本保持不变。以上结果表明,通过改变催化剂种类和催化剂的量可以对反应进程进行调控。

Scheme 1 表面修饰示意图

图1 催化剂化学结构

图2 (a)不同催化剂条件下接触角变化曲线;(b)不同吡啶二甲酸浓度下接触角变化曲线

图3 吡啶二甲酸作催化剂条件下的(a)接触角照片和(b)接触角变化趋势
3.2 表面反应动力学测定
利用静态水接触角的方法同样可以对吡啶二甲酸催化ab-NTA和表面VS基团反应的反应动力学进行测定。在此选择了 10%吡啶二甲酸作为催化剂的条件进行研究。通过测量不同反应时间的表面静态水接触角,再利用Cassie公式计算可以得到表面覆盖度23:

其中 χcarboxyl和 χVS分别表示羧基的表面覆盖度以及相对于羧基VS基团的表面覆盖度。
由接触角照片(如图 3a)可以明显看出反应前后表面接触角变化。图3b为吡啶二甲酸作催化剂条件下金表面接触角的变化曲线,结果显示当表面为VS基团时疏水性较强接触角为66.1° ± 0.6°,随着反应的进行,反应24 h后接触角降为42.9° ±0.5°。经测量利用羧基硫醇自组装得到的羧基表面接触角为 θcarboxyl= 37.2° ± 1°。由于每个 NTA 分子含有三个羧基,通过换算可以得到不同反应时间VS基团的表面覆盖度。将VS基团的表面覆盖度取自然对数与时间作图得到图4,可以看出VS表面覆盖度的自然对数与时间呈线性相关,这表明这一表面反应可以用一级反应动力学进行描述。反应的速率常数和半衰期可以表示为:

其中χVS和χVS0分别表述VS测量的和最初的表面覆盖度,t和t0分别表示测量时间和最初时间,kobs为反应速率常数,t1/2表示反应半衰期。
通过计算得到吡啶二甲酸催化表面 VS基团与ab-NTA氨基的反应速率常数为0.0012 min−1,半衰期为569 min。从计算结果看出,该反应较为缓慢,易于通过调控反应时间对配基表面密度进行调控。

图4 VS (乙烯砜基)基团表面覆盖度的自然对数
3.3 表面膜电位和XPS表征
为了对修饰前后表面进行表征,分别利用表面膜电位和XPS的方法对VS表面和ab-NTA表面进行了检测。表面膜电位方法通过对表面电位变化进行检测,可以反映出表面基团的变化。
如图 5所示为不同表面的表面膜电位,裸金表面的面电位是−28.33 mV,这是由于金表面含有大量自由电子造成的。当表面修饰上乙烯基砜二硫化化合物后变为−5.45 mV,吡啶二甲酸催化表面VS与ab-NTA反应12 h后,由于表面羧基增多,表面电位降为−15.33 mV。表面膜电位的变化表明了VS二硫化合物的自组装和ab-NTA与VS基团的反应均成功进行。为进一步对表面结构进行表征,我们对 VS表面和 ab-NTA表面进行了 XPS表征。结果如图6所示为XPS谱图,从S 2p谱呈现出两个峰,分别裂分为峰面积1 : 2的双峰,其中163 eV周围的峰归属为S-Au峰,169 eV周围的峰归属为砜基峰,这表明VS二硫化合物成功组装到了金表面。VS表面的C 1s谱在进行拟合后可以分为两部分:285 eV处的峰归属为亚甲基链的峰,另一处285.6 eV处的峰归属为与砜基相连的碳原子的峰。与VS表面相比ab-NTA表面的C 1s谱在287 eV和288.5 eV处出现两个峰,分别可以归属为与亚胺相连的碳原子的峰和羧基上碳原子的峰。由两种表面的N 1s谱可以看出,VS表面没有检测到N元子,ab-NTA表面在400.4 eV处有N元素的峰,可以归属为亚胺的峰。结果表明ab-NTA与表面VS基团成功发生了反应。

图5 表面膜电位

图6 (1) VS表面和(2) ab-NTA表面的XPS表征结果
3.4 ab-NTA表面生物功能表征
NTA基团在与镍离子螯合之后可以与组氨酸上的咪唑残基发生配位,从而可以特异性结合组氨酸标签的蛋白24。因此通过SPR对NTA表面的蛋白质结合动力学和热力学进行研究,可以对表面 NTA密度进行表征。本文选用的蛋白是SA-6His。以NHS活化羧基的方法25,26作为对照。
基于VS和NHS的ab-NTA表面均反应12 h以保证充分反应。如图7所示为基于VS的ab-NTA表面在通入不同浓度蛋白时的SPR曲线。从图中可以看出随着蛋白浓度的升高,其结合量呈现明显的梯度增高。在通过咪唑洗脱和PBS洗涤之后能够回到基线,表面基本没有蛋白质残留,这表明 SA-6His蛋白与表面的结合为特异性结合。在通过0.25 mg·mL−1蛋白时可以看到蛋白结合量首先迅速增高,随后出现缓慢降低的现象。我们推测,在通入高浓度蛋白时,蛋白与表面NTA首先形成了单价态结合,随着蛋白溶液的持续通过,蛋白与NTA逐渐转变为更稳定的多价态结合,结合量随之有所降低。如图8所示,选择通蛋白30 min时的响应值作为该浓度下的结合量,利用Langmuir方程对数据进行拟合,得到了NTA表面对 SA-6His和 BSA的等温吸附曲线(图 8),并与NHS方法进行对比。通过计算可以得到了两种表面的 Kd和 Rmax(表 1)。
对于 SA-6His蛋白的等温吸附,基于 VS的NTA 表面 Kd为 0.60 × 10−6mol−1,基于 NHS 的 NTA表面 Kd为 1.01 × 10−6mol−1,由此可以看出基于 VS的NTA表面对于SA-6His蛋白的结合强度更高。从最大结合量来看,基于VS的NTA表面的最大结合量为693,基于NHS的NTA表面的是457。
以上结果表明,基于VS的NTA表面修饰方法较基于NHS的NTA表面修饰方法能够提供更高的结合强度和结合量,说明相对于NHS的方法基于 VS的表面修饰方法可以提供更高的配基密度。由图8也可以看出两种表面对BSA的非特异性吸附均很低,表明基于VS的方法同样具有较好的抗非特异性吸附能力。
由于NHS酯在碱性水溶液中易水解,导致反应过程伴随着水解使得基于NHS方法的配基表面密度难以提高。而基于VS的反应符合一级反应动力学,最终可以达到较高的反应效率,所以能够提高供更高的配基表面密度。与NHS方法相比,基于 VS的方法反应效率更高,配基表面密度更高,并且具有很好抗非特异性吸附能力。

图7 蛋白质与VS-ab-NTA表面的SPR结合曲线
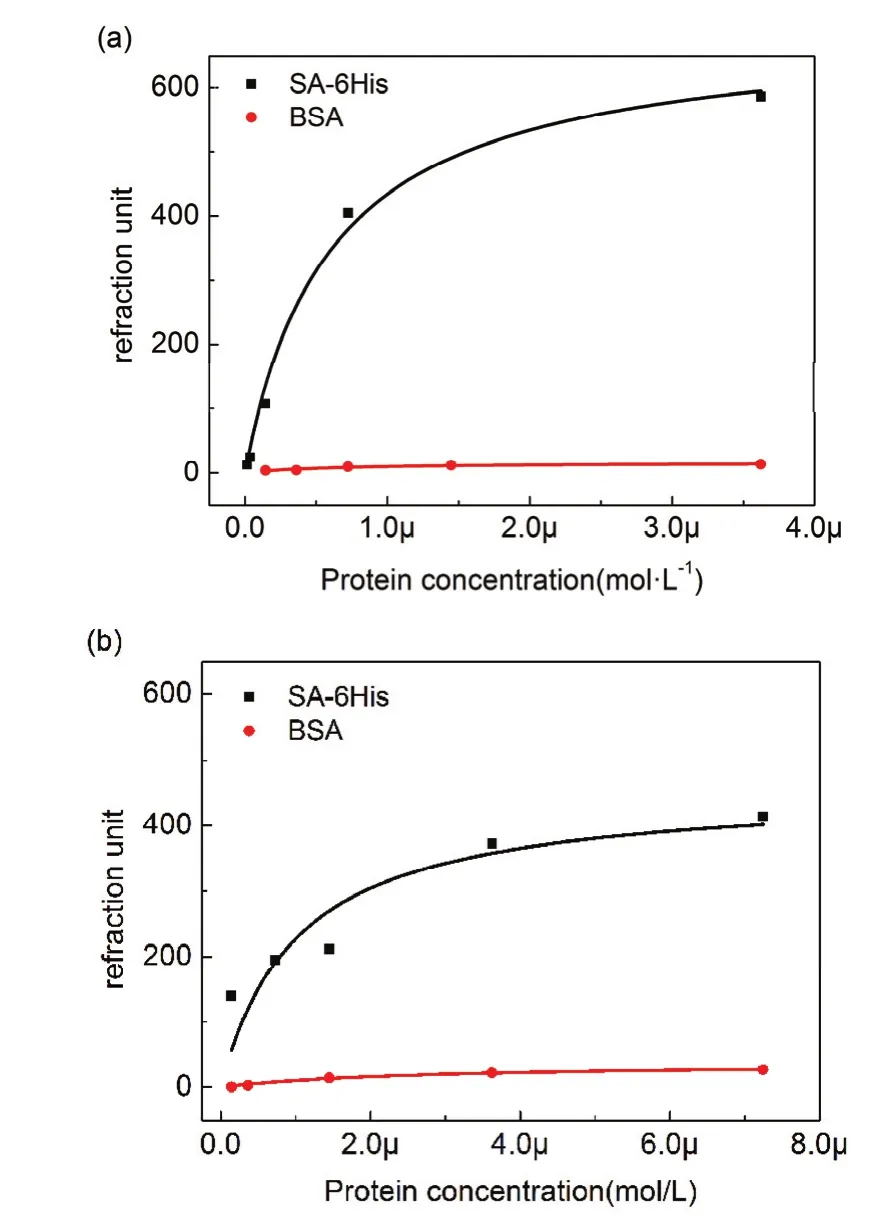
图8 基于 VS (a)和 NHS (b)的ab-NTA表面的等温吸附曲线

表1 基于VS和NHS的ab-NTA表面的Kd和RmaxTable 1 Kd and Rmax of ab-NTA surface base on VS and NHS.
3.5 NTA配基密度控制
结合上文催化剂筛选以及反应动力学结果,分别利用吡啶二甲酸催化0.5、2、12 h以及利用三苯基膦催化12 h得到四种不同ab-NTA密度的SPR芯片。利用 SPR对四种芯片分别进行了SA-6His蛋白的静态吸附实验。如图9 (Cat.1)所示为吡啶二甲酸作为催化剂催化不同时间得到的三种不同配基密度的芯片的静态吸附曲线,从图中可以看出种不同催化时间芯片的静态吸附曲线呈现明显的梯度。图9中12 h (Cat.2)为三苯基膦作为催化剂催化12 h表面的静态吸附曲线,与图9 12 h (Cat.1)曲线对比可以看出,两个静态吸附曲线具有明显梯度。上述结果再次证明通过控制反应时间和催化剂种类能够对配基表面密度进行调控。
利用上文反应动力学结果可以计算出四种芯片表面羧基的表面覆盖度分别为14.5%、41.2%、80.6%和34.5%。根据报道羧基硫醇自组装膜的密度为6.98 molecule nm−227。通过估算可以得出四种表面的ab-NTA密度分别约为0.34、0.96、1.88和 0.80 molecule·nm−2。利用 Langmuir方程对数据进行拟合后可以到的表 2所示结果,四种不同配基密度表面的蛋白最大结合量分别为183、504、693和282,可以看出蛋白最大结合量与配基表面密度呈正相关,随着配基密度的增加,表面最大结合量增大。
表 2结果显示四种不同配基密度表面对SA-6His蛋白的 Kd值分别为 1.13 × 10−6、0.62 ×10−6、0.60 × 10−6和 0.58 × 10−6mol−1。可以看出吡啶二甲酸催化0.5 h的表面配基与SA-6His蛋白的结合强度为 1.13 × 10−6mol−1,明显低于另外三种表面。由表面ab-NTA密度可以估算得到四种表面的NTA分子平均间距分别约1.7、1.0、0.7和1.1 nm。组氨酸标签与NTA的结合属于多价态结合28,每两个咪唑基团可以与一个镍离子配位,六个组氨酸标签的长度约为2.0 nm。对比以上数据可以看出,在低密度表面上每个组氨酸标签蛋白只能与1–2个NTA分子络合,此时蛋白与NTA之间多数形成单价态结合,所以结合强度较低。吡啶二甲酸催化2、12 h以及三苯基膦催化12 h的表面上每个蛋白则能够与2–3个NTA分子络合,可以形成多价态相互作用,结合强度明显提高。
上述实验结果通过控制表面 VS基团与氨基的催化反应时间以及催化剂种类实现了配基表面密度调控,并建立了表面密度与性能之间的构效关系。该工作为表面密度控制提供了新的方法。对于生物传感器修饰、蛋白纯化配基密度优化、生物医学材料表面修饰等研究工作,利用本文的配基表面密度控制方法可以在表面模拟不同配基密度的材料表面环境,结合SPR、QCM等研究方法可以简便地不同配基密度表面的特异性/非特异性结合性能进行表征,提高了密度筛选的效率。
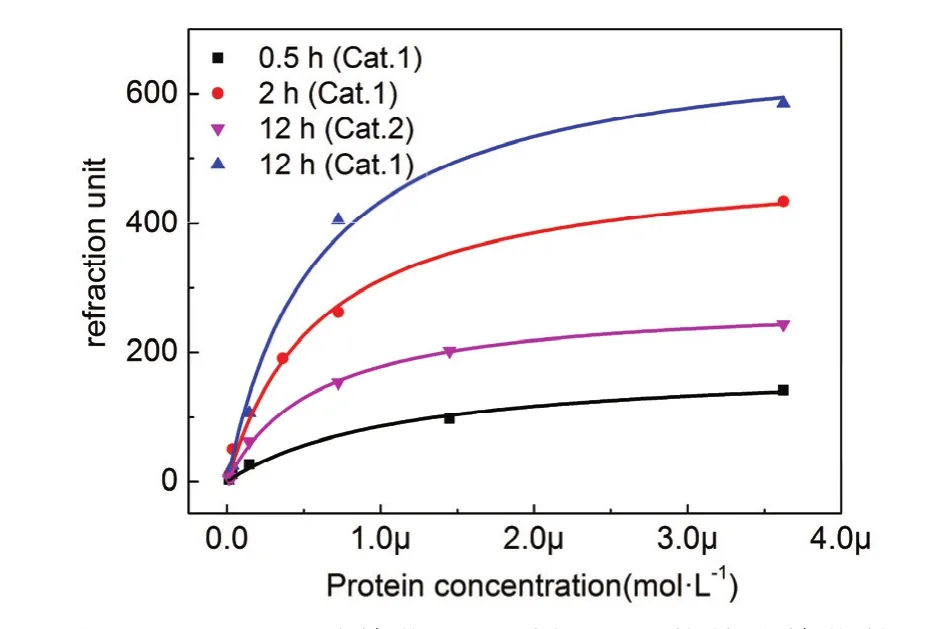
图9 吡啶二甲酸催化不同时间和三苯基膦催化的表面的等温吸附曲线

表2 不同反应时间和催化剂催化的ab-NTA表面的Kd和RmaxTable 2 Kd and Rmax of ab-NTA surface with different reaction time and catalysts.
4 结论
本文利用静态水接触接触角的方法对一系列N、P中心的催化剂催化表面VS基团与氨基反应的催化性能进行了表征,结果显示吡啶二甲酸作为表面VS与氨基反应的催化剂具有最高的催化效率,DMAP、一甲基咪唑和三苯基膦也可以催化该反应。对吡啶二甲酸催化反应动力学的研究结果显示该反应符合一级反应动力学,其反应速率常数为0.0012 min−1,半衰期为569 min。本文以ab-NTA作为配基,分别利用传统的NHS活化法和VS法对金表面进行了修饰,并利用SPR技术对表面蛋白静态吸附进行了研究,结果表明利用VS的方法能够提供更高的配基表面密度。根据反应动力学结果分别利用不同催化剂对 VS表面进行不同时间的修饰,并结合SPR对不同反应时间的表面进行了蛋白质静态结合实验,结果表明通过控制反应时间和催化剂种类均可以实现配基表面密度控制。同时,在静态结合实验中观察到了单价态相互作用和多价态相互作用。反应0.5 h的表面由于配基密度低,配基分子间距大,造成蛋白质与配基的结合倾向于单价态结合,结合强度较高密度表面有明显的降低。
本文所发展的催化剂催化的表面乙烯砜基与氨基的反应能够高效地在表面引入含有氨基的分子,并能够通过控制反应时间和催化剂种类对表面反应进程进行控制,从而实现对配基表面密度的调控,为表面密度控制提供了新方法。与混合自组装方法相比本方法可控性强、应用广泛并且具有一定普适性。
(1) Yuan, P. X.; Deng, S. Y.; Yao, C. G.; Wan, Y.; Cosnier, S.; Shan, D.Biosens. Bioelectron. 2017, 89, 319. doi: 10.1016/j.bios.2016.07.031
(2) Cabanas-Danes, J.; Rodrigues, E. D. J. Am. Chem. Soc. 2014, 136,12675. doi: 10.1021/ja505695w
(3) Nakamura, I.; Horikawa, Y.; Makino, A.; Sugiyama, J.; Kimura, S.Biomacromolecules 2011, 12, 785. doi: 10.1021/bm101394j
(4) Schartner, J.; Hoeck, N. Anal. Chem. 2015, 87, 7467.doi: 10.1021/acs.analchem.5b01823
(5) Cheng, F.; Li, M. Y.; Wang, H. Q.; Lin, D. Q.; Qu, J. P. Langmuir 2015, 31, 3422. doi: 10.1021/la5044987
(6) Rowley, J. A.; Mooney, D. J. J. Biomed. Mater. Res. 2002, 60, 217.doi: 10.1002/jbm.1287
(7) Shoffstall, A. J.; Everhart, L. M. Biomacromolecules 2013, 14, 2790.doi: 10.1021/bm400619v
(8) Chen, X. W.; Pei, D. H. J. Comb. Chem. 2009, 11, 604.doi: 10.1021/cc9000168
(9) Shao, Q.; Jiang, S. Y. J. Phys. Chem. B 2014, 118, 7630.doi: 10.1021/jp5027114
(10) Tomohiro, H.; Kenji, W. J. Phys. Chem. C 2009, 113, 18795.doi: 10.1021/jp906494u
(11) Subramanian, A.; Irudayaraj, J.; Ryan, T. Sensor. Actuat. B:Chem. 2006, 114, 192. doi: 10.1016/j.snb.2005.04.030
(12) Ma, H.; Wells, M.; Beebe, T. P. Jr.; Chilkoti, A. Adv. Funct.Mater. 2006, 16, 640. doi: 10.1002/adfm.200500426
(13) Bain, C. D.; Whitesides, G. M. J. Am. Chem. Soc. 1988, 110,6560.
(14) Bohmler, J.; Ponche, A.; Anselme, K.; Ploux, L. ACS. Appl.Mater. Inter. 2013, 5, 10478. doi: 10.1021/am401976g
(15) Tomohiro, F.; Yoshiko, M. Bioconjugate Chem. 2010, 21,1079. doi: 10.1021/bc100053x
(16) Liu, Y. T.; Yan, L.; Sun, L. M.; Li, H. Q.; Li, H. H. Chem. Eng.(China) 2014, 42, 69. [刘玉婷, 颜莉, 孙立民, 李慧琴, 李海华. 化学工程, 2014, 42, 69.]doi: 10.3969/j.issn.1005-9954.2014.03.014
(17) Cheng, F.; Wang, H. Q.; Xu, K.; He, W. Acta Phys. -Chim.Sin. 2017, 33, 426. [程昉, 王汉奇, 许旷, 何炜. 物理化学学报, 2017, 33, 426.] doi: 0.3866/PKU.WHXB201609291
(18) Eugene W. L.; Chan, M. N. Y. J. Am. Chem. Soc. 2006, 128,15542. doi: 10.1021/ja065828l
(19) Zhang, S.; Maidenberg, Y.; Luo, K.; Koberstein, J. T.Langmuir 2014, 30, 6071. doi: 10.1021/la501233w
(20) Wang, H. Q.; Cheng, F.; Li, M. Y.; Peng, W.; Qu, J. P.Langmuir 2015, 31, 3413. doi: 10.1021/la504087a
(21) Esteves, A. P.; Silva, M. E.; Rodrigues, L.M.;Oliveira-Campos, A. M. F.; Hrdina, R. Tetrahedron Lett. 2007,48, 9040. doi: 10.1016/j.tetlet.2007.10.077
(22) Wang, C.; Qi, C. Z. Tetrahedron 2013, 69, 5348.doi: 10.1016/j.tet.2013.04.123
(23) Cassie, A. B. D.; Baxter, S. Trans. Faraday Soc. 1944, 40,546.
(24) Kim, E. J.; Chung, B. H.; Lee, H. J. Anal. Chem. 2012, 84,10091. doi: 10.1021/ac302584d
(25) Maalouli, N.; Gouget-Laemmel, A. C. Langmuir 2011, 27,5498. doi: 10.1021/la2005437
(26) Pei, J.; Tang, Y.; Xu, N.; Lu, W.; Xiao, S. J.; Liu, J. N. Sci.China Chem. 2010, 54, 526. doi: 10.1007/s11426-010-4128-3
(27) Shin-ichiro, I.; Takashi, K. J. Electroanal. Chem. 1997, 428,33. doi: 10.1016/S0022-0728(97)00006-5
(28) Suman L.; Jacob, P. J. Am. Chem. Soc. 2005, 127, 10205.doi: 10.1021/ja050690c
