磷酸钇晶体物理性质及本征缺陷的研究
2018-02-03,,,
, , ,
(上海理工大学 理学院,上海 200093)
磷钇矿由于其在核能领域、光子器件和光学通讯上的广泛应用得到了越来越多的关注[1].磷酸钇(YPO4)与独居石类似,都是常见的化学性质稳定的氧化物结构,具有高熔点和耐腐蚀性的特性[2],也是天然含有大量铀和钍的正磷酸盐,又经历了20亿年时间暴露在α粒子和相关反冲核的环境中,使得磷钇矿的结晶结构尤其是磷原子与氧原子构成的四面体具有很强的稳定性,所以,磷钇矿材料被认为是处理核废料,特别是高放废料的主要物质[3].
磷钇矿纯净物的经济制备为研究磷酸钇掺杂不同浓度稀土系列的发光材料打下了坚实的基础[4].近几年来,用水溶凝胶法[5]、共同沉淀法和燃烧法等方法[6]合成后的稀土磷酸钇材料的荧光性能大大提高,也开展了对晶体磷酸钇掺杂稀土离子后的纳米晶体的发光方面的研究[7].晶体磷酸钇随着稀土离子掺杂浓度的不同可以表现出闪烁性质[8-9],而且掺杂稀土钕离子后,其衰减时间最快,为6 ns[10].但是,有关磷酸钇晶体物理性质和本征缺陷的研究报道还不多,为了提高磷酸钇晶体掺杂稀土离子后的发光效率,需对完整磷酸钇晶体的基本性质进行研究.本文利用基于分子动力学原理的GULP软件并参考已有的3组势参数对晶体YPO4的物理性质和本征缺陷展开研究,这些研究结果将为后续的研究提供理论基础.
1 计算方法
稀土钇离子(Y3+)半径较小,形成的YPO4晶体具有磷钇矿结构,属于四方晶系(a=b=0.688 17 nm,c=0.601 77 nm),空间群为I41/amd.a,b,c为晶格参数.在这个结构中,每个Y3+被8个O2-包围,形成对称性为D2d的十二面体结构[YO8],这8个O2-因Y—O键键长不同分为两组,一组为Y—O键较长的,这4个O2-分别与周围的四面体PO4相连,另一组Y—O键较短的,4个O2-分别和十二面体[YO8]相连[11-12],如图1所示.
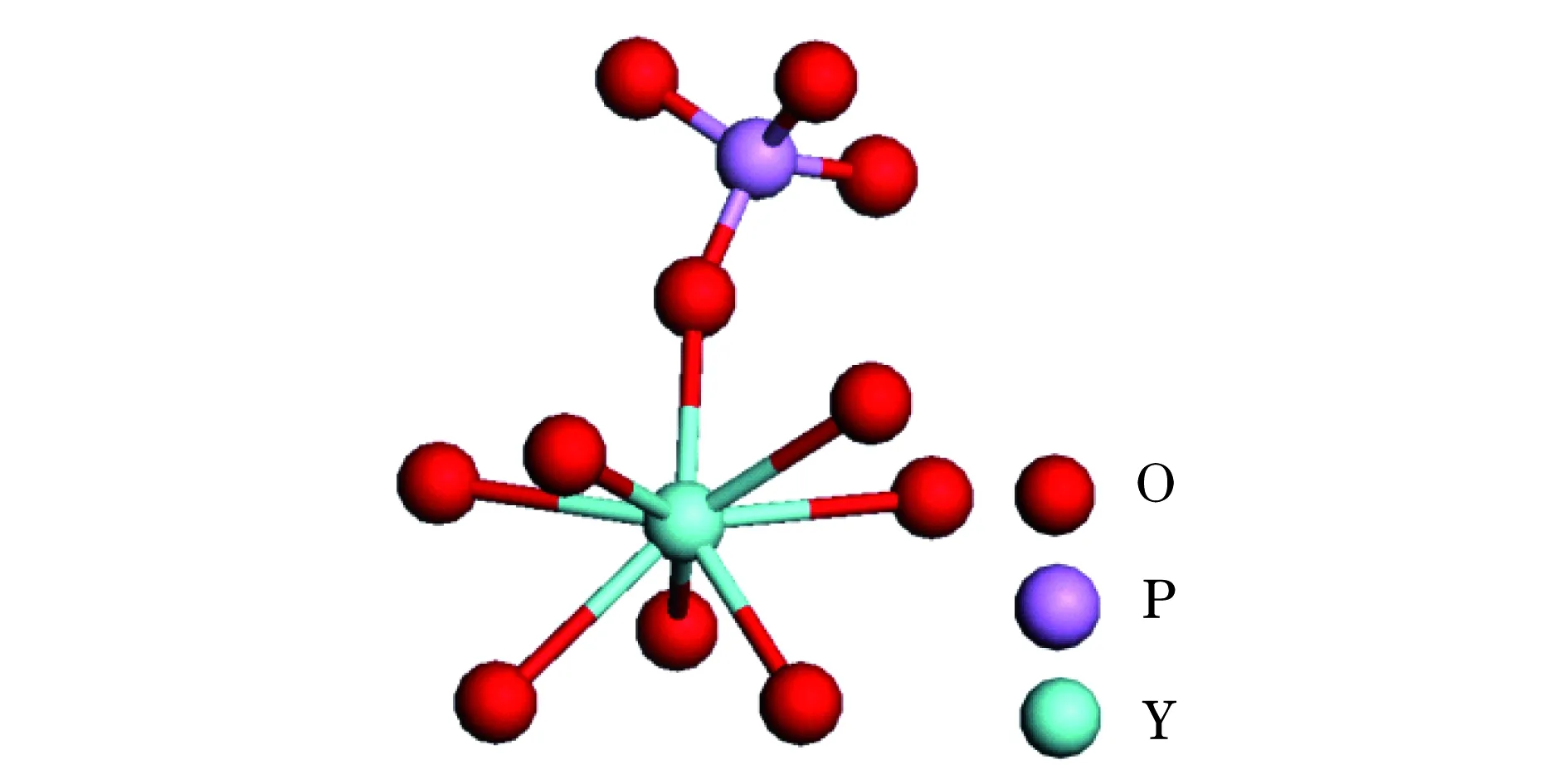
图1 [YO8]的示意图Fig.1 Sketch map of the structure of [YO8]
本文采用基于静态原子晶格计算的软件GULP (general utility lattice program)[13-14],该软件是在玻尔(Born)离子晶格的模型上,为了大大降低计算机的运算时间,将点缺陷周围的晶格用Mott-Littleton法简化成3个区域,分别为 Ⅰ,Ⅱa,Ⅱb[15].再利用势参数模拟计算完整磷酸钇晶体的力学性质和热学性质.其中,区域 Ⅰ 采用原子水平处理,所有的离子由于缺陷的存在会发生弛豫,直到重新达到平衡为止.而区域 Ⅱ 因与缺陷位置的距离较远,受到缺陷的作用力较小,所以,采用准连续介质的方法模拟缺陷周围离子的扩散.
使用GULP软件模拟计算YPO4晶体的物理性质和本征点缺陷的缺陷形成能时,最常用的势参数是描述晶体原子间的相互作用势Uij.
(1)
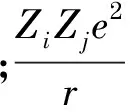
由于YPO4晶体中有共价键存在,所以,原子之间有排斥作用,甚至有孤对电子产生,此时可以用three-body势Vijk来简化计算.
(2)
式中:θijk为计算优化后O—P—O键和P—O—P键的键角;θ0为计算优化前O—P—O键和P—O—P键的键角;kijk为可调节的势参数.
除了Bucking-ham势,也可以用Morse势进行计算,Morse势可以隐含键断裂这种现象,对于分子振动的微细结构计算具有良好的近似作用.Morse势
(3)
式中:D为原子之间的键断开所需的能量;r为计算优化后原子i与原子j之间的距离;r0为计算优化前原子间的键长;α为可调节的势参数.
为了得到磷钇矿的基本性质,选取3组磷酸钇晶体原子间的相互作用势进行计算,如表1所示(见下页).“磷钇矿1”的相互作用势选自文献[16],钇离子(Y3+)、磷离子(P5+)和氧离子(O2-)的带电数分别为1.8 e,3.0 e和-1.2 e.“磷钇矿2”的相互作用势选自文献[3],钇离子、磷离子和氧离子的带电数分别为2.3 e,2.7 e和-1.25 e.“磷钇矿3”的相互作用势选自文献[3],钇离子、磷离子和氧离子的带电数分别为1.4 e,3.4 e和-1.2 e.
YPO4晶体中原子会在其位置上下振动,是热运动现象,即晶体的热学性质主要由晶格振动决定.一般情况下,晶体的内能由晶格振动的能量和电子运动能量两部分组成,这两部分能量都会受到温度的影响,因此,晶体的内能对YPO4晶体的比热容也有着不可忽略的影响.当温度不是很低时,晶格振动的能量对晶体的热作用逐渐表现出来.YPO4晶体的本征点缺陷的振动熵
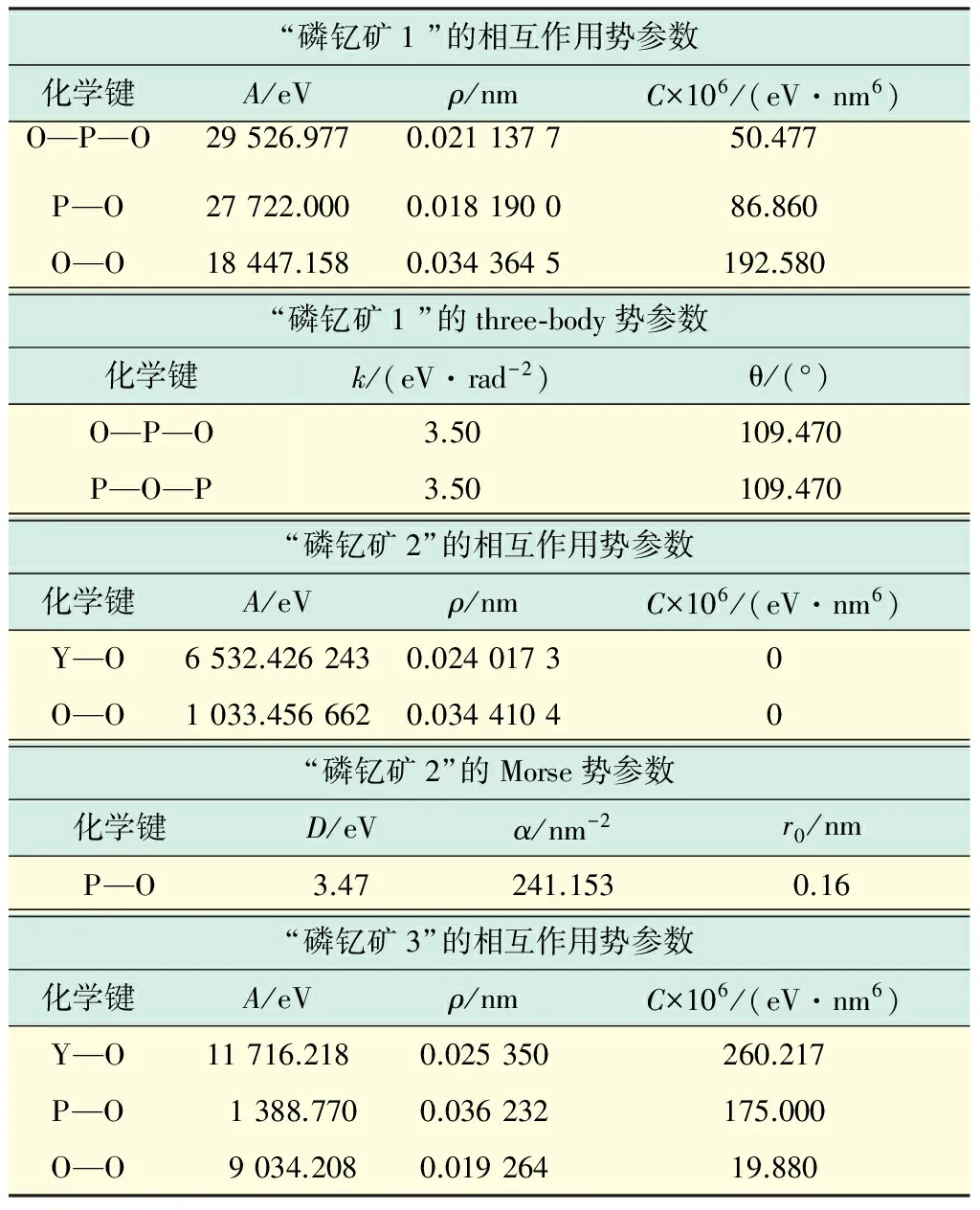
表1 3组用于计算磷酸钇晶体物理性质和缺陷形成能的势参数Tab.1 Three sets of parameters of interatomic potentials
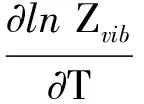
(4)
(5)
式中:Zvib为振动熵的配分函数;R,k为常数;T为温度;wk为角频率;hv为光子能量.
模拟计算YPO4晶体本征点缺陷的缺陷形成能时,可以找到在晶体中占主导作用的缺陷[17].缺陷形成能的计算公式为
E=E1-E2+E
(6)
式中:E1为完整的YPO4晶体中有点缺陷时的总能量;E2为完整YPO4晶体的总能量;E∞为有缺陷时引入的校正能量[18].本文计算对象为磷酸钇晶体(YPO4)的本征点缺陷,有氧空位、磷空位、钇空位、氧填隙、磷填隙、钇填隙、反位钇,反位磷和钇、磷、氧原子的弗伦克尔缺陷.
2 计算结果与讨论
2.1 YPO4晶体结构参数的计算结果
为了验证所选取的势参数的可靠性,用“磷钇矿1”,“磷钇矿2”和“磷钇矿3”的势参数对YPO4晶体进行结构优化计算,计算得出的晶格常数、键长、键角和体积的结果与实验结果较符合,如表2所示.V为体积.从表2中可以看出,计算结果与实验结果间的最大误差小于5%,所以,文献[16]和文献[3]给出的势参数较为合理.
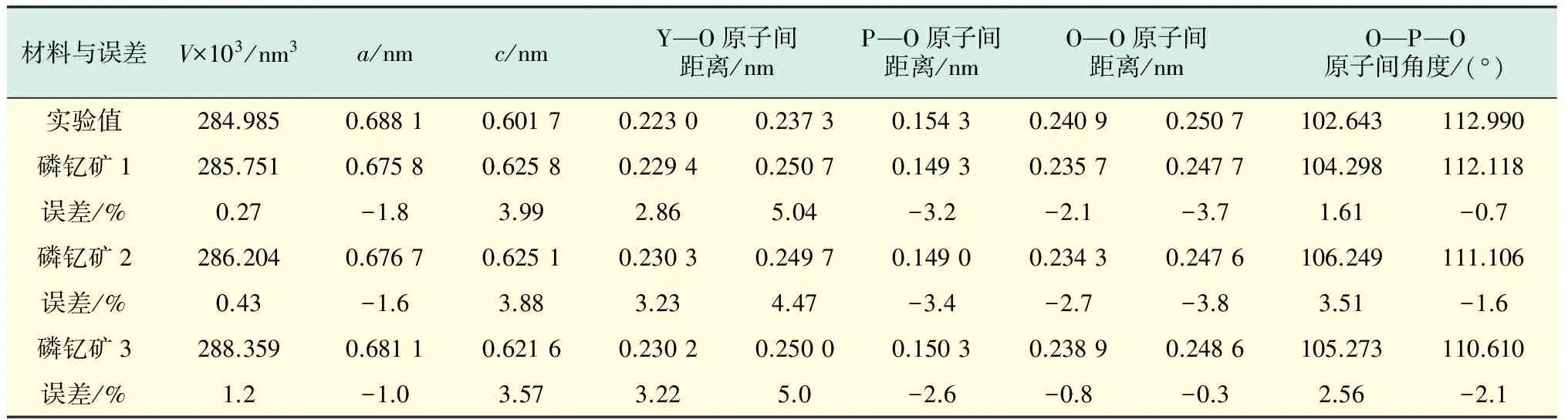
表2 YPO4晶体的晶体结构优化前后的比较Tab.2 Comparison of initial and final structures of YPO4 crystal
2.2 YPO4晶体的力学性质
力学性质中的弹性常数作为表征材料性能的主要参数,可以反映晶体形变的机械硬度.利用“磷钇矿1”,“磷钇矿2”和“磷钇矿3”的势参数进行计算,将弹性常数C11~C66的结果与实验值比较,如表3 所示.弹性常数C11和C33分别表示YPO4晶体在a轴和c轴方向的抵抗应变能力,即YPO4晶体在(001)和(100)方向上的化学键的强度.由表3数据可知,YPO4晶体在c轴方向的抗应变能力最强,这是与实验值吻合的[19],而C11和C66存在的误差是由于计算简便忽略了离子极化的影响.

表3 YPO4 晶体的弹性常数计算值与实验值Tab.3 Calculated and experimental values of the elastic constants of YPO4 crystal GPa
2.3 YPO4晶体的热学性质
利用已有的3组势参数对晶体的本征点缺陷进行计算,包括氧空位VO、磷空位VP、钇空位VY、氧填隙Oi、磷填隙Pi、钇填隙Yi、反位钇YP、反位磷PY.由于缺陷的存在会改变周围原子的振动频率,会引起振动熵的改变.图2给出了不同温度下振动熵(TΔS)对“磷钇矿1”,“磷钇矿2”和“磷钇矿3”的本征点缺陷形成能的影响.由于“磷钇矿1”,“磷钇矿2”和“磷钇矿3”的势参数选取不同,振动熵对晶体的本征点缺陷形成能会随着温度的变化而变化,但整体趋势相同.填隙类缺陷如氧填隙Oi、磷填隙Pi、钇填隙Yi,这3种缺陷的振动熵对缺陷形成能的作用随着温度的上升,沿负方向增大;而空位类缺陷如氧空位VO,磷空位VP,钇空位VY,这些缺陷的振动熵对缺陷形成能的作用随着温度上升,沿正方向增大,这些现象在物理学领域是合理的.图2表明,对于“磷钇矿1”,YP的振动熵随温度变化幅度较小;对于“磷钇矿2”,Pi的振动熵随温度变化较小;对于“磷钇矿3”,YP的振动熵随温度的变化较小.一般情况下,振动熵对缺陷的作用是比较小的,是可以忽略的,但计算结果表明:在高温条件下,振动熵对YPO4晶体的本征点缺陷形成能的影响比较大,是不能忽略的[20].
2.4 YPO4晶体的本征点缺陷
利用已有的势参数,计算晶体YPO4的本征点缺陷的形成能,计算结果如表4所示(见下页).在计算孤立填隙原子的缺陷形成能时,要找到其最佳的填隙位置,使其在此位置形成的缺陷形成能最低.此次计算寻找了几处填隙位置,但计算结果表明,缺陷具有最小的缺陷形成能的位置是一样的,填隙原子会与周围的2个磷原子和4个氧原子构成八面体.从表4中可以看出,磷填隙的缺陷形成能最小,反位磷的缺陷形成能也较小.但孤立的点缺陷形成是没有意义的,P—O键是共价键,磷又与两类氧原子形成的四面体具有强的稳定性,所以,较Y—O键需要很高的能量才能断裂,这也就解释了磷原子(P)的弗伦克尔缺陷很难形成.本文对钇原子和氧原子的弗伦克尔缺陷形成能的计算结果表明:在晶体YPO4中氧空位和氧的弗伦克尔缺陷的缺陷形成能是最低的,相对其他缺陷更容易形成,同时氧原子的弗伦克尔缺陷是该晶体的主要缺陷.
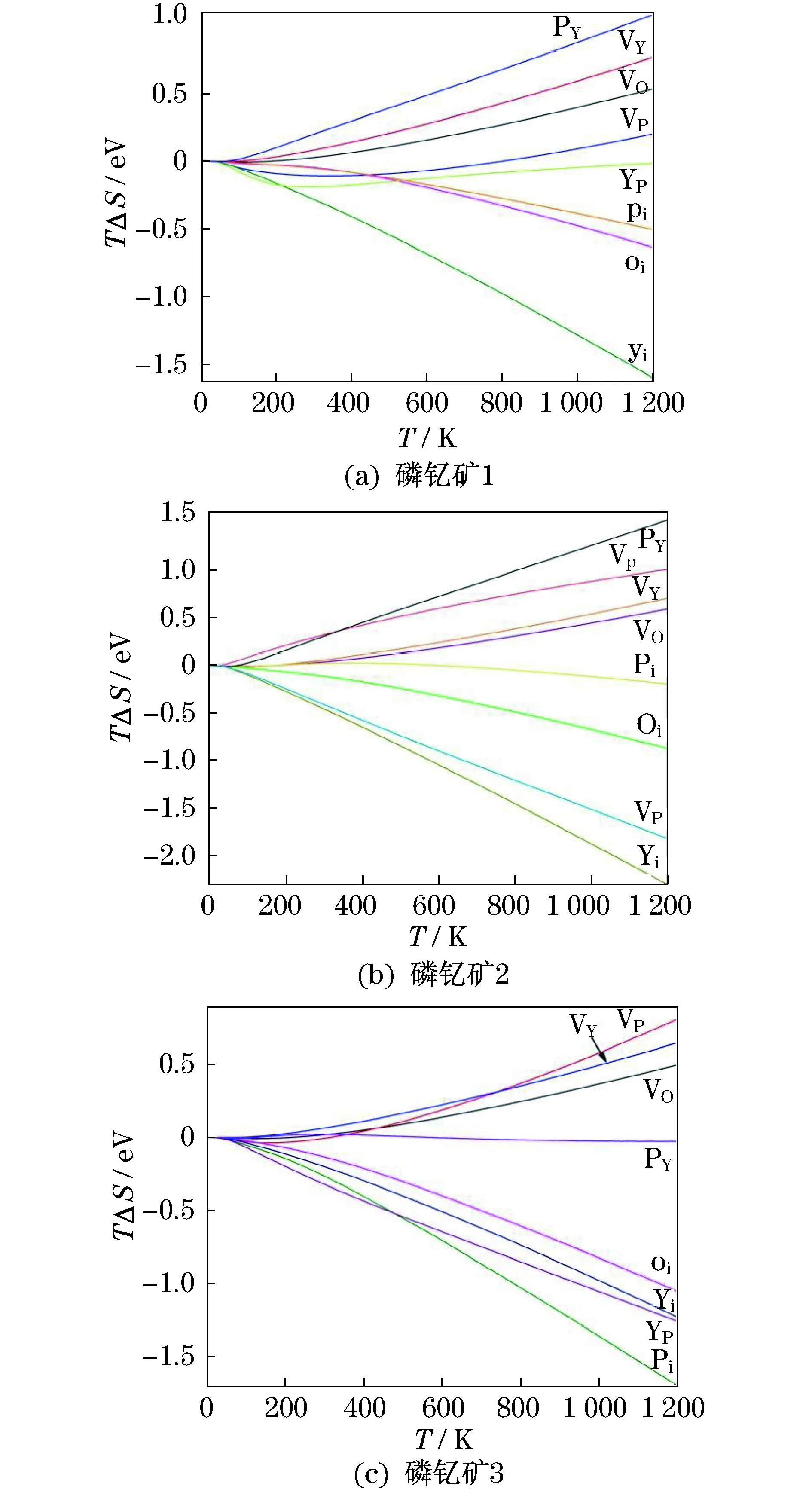
图2 振动熵在不同温度下对“磷钇矿1”,“磷钇矿2”和“磷钇矿3”的本征点缺陷形成能的影响
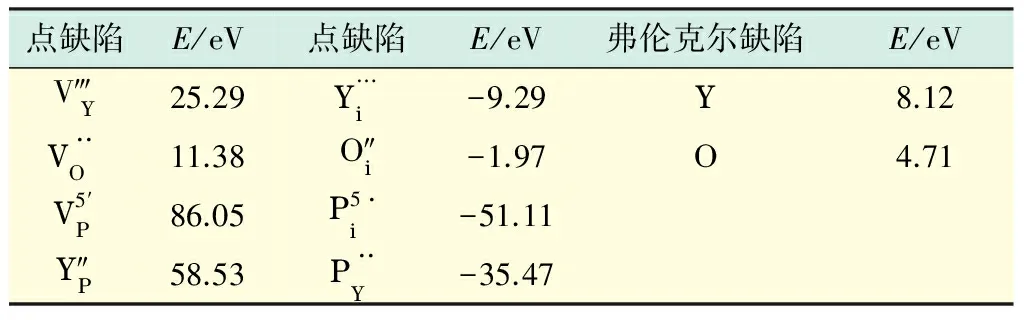
点缺陷E/eV点缺陷E/eV弗伦克尔缺陷E/eVV‴Y25.29Y…i-9.29Y8.12V··O11.38O″i-1.97O4.71V5′P86.05P5·i-51.11Y″P58.53P··Y-35.47
3 结 论
采用3组不同的势参数,通过计算得到磷酸钇晶体的结构参数、弹性常数和本征点缺陷的振动熵.采用3组势参数计算得到的YPO4晶体结构参数与实验值较吻合.计算得到的YPO4晶体的弹性常数表明,晶体YPO4在(100)方向的抗应变能力是最强的.由晶体YPO4的本征点缺陷振动熵的结果了解到:振动熵对本征点缺陷形成能的影响是不能忽略的,尤其是在高温条件下.从模拟计算YPO4晶体的本征点缺陷的缺陷形成能结果可知,氧空位和氧的弗伦克尔缺陷在晶体中相对其他缺陷更容易形成.
[1] LAI H,BAO A,YANG Y M,et al.UV luminescence property of YPO4:RE (RE=Ce3+,Tb3+)[J].The Journal of Physical Chemistry C,2008,112(1):282-286.
[2] RAMBABU U,MUNIRATHNAM N R,PRAKASH T L,et al.Emission spectra of LnPO4:RE3+(Ln=La,Gd; RE=Eu,Tb and Ce) powder phosphors[J].Materials Chemistry and Physics,2003,78(1):160-169.
[3] URUSOV V S,GRECHANOVSKY A E,EREMIN N N.Radiation resistance of the xenotime YPO4from the computer simulation data[J].Glass Physics and Chemistry,2012,38(1):55-62.
[4] 王达君.稀土离子系列掺杂的晶体的电子结构和4f→5d激发能的计算[D].合肥:中国科学技术大学,2009.
[5] KAHOUADJI B,GUERBOUS L,LAMIRI L,et al.Structural and optical properties of Ce3+doped YPO4:nanophosphors synthesis by sol gel method[J].International Scholarly and Scientific Research & Innovation,2014,8(4):683-641.
[6] DU G H,KAN X Y,HAN Y B,et al.Structure and luminescence of YPO4:Dy3+microflowers[J].Materials Letters,2012,74:229-231.
[7] HUANG J S,GAO R,LU Z G,et al.Sol-gel preparation and photoluminescence enhancement of Li+and Eu3+co-doped YPO4nanophosphors[J].Optical Materials,2010,32(9):857-861.
[8] WISNIEWSKI D,TAVERNIER S,DORENBOS P,et al.VUV scintillation of LuPO4:Nd and YPO4:Nd[J].IEEE Transactions on Nuclear Science,2002,49(3):937-940.
[9] WISNIEWSKI D,TAVERNIER S,WOJTOWICZ A J,et al.LuPO4:Nd and YPO4:Nd-new promising VUV scintillation materials[J].Nuclear Instruments and Methods in Physics Research Section A:Accelerators,Spectrometers,Detectors and Associated Equipment,2002,486(1/2):239-243.
[10] WANG D W,HUANG S H,YOU F T,et al.Scintillation properties of YPO4:RE (RE=Ce3+,Pr3+or Nd3+)[J].Chinese Physics C,2009,33(11):1019-1022.
[11] LIU Q,SU Y G,YU H S,et al.YPO4nanocrystals:preparation and size-induced lattice symmetry enhancement[J].Journal of Rare Earths,2008,26(4):495-500.
[12] DI W H,WANG X J,CHEN B J,et al.Preparation,characterization and VUV luminescence property of YPO4:Tb phosphor for a PDP[J].Optical Materials,2005,27(8):1386-1390.
[13] DICK Jr B G,OVERHAUSER A W.Theory of the dielectric constants of alkali halide crystals[J].Physical Review,1958,112(1):90-103.
[14] GALE J D.GULP:a computer program for the symmetry-adapted simulation of solids[J].Journal of the Chemical Society,Faraday Transactions,1997,93(4):629-637.
[15] GRIMES R W,CATLOW C R A,STONEHAM A M.Quantum-mechanical cluster calculations and the Mott-Littleton methodology[J].Journal of the Chemical Society,Faraday Transactions,1989,85(5):485-495.
[16] GAO F,XIAO H Y,ZHOU Y G,et al.Ab initio study of defect properties in YPO4[J].Computational Materials Science,2012,54:170-175.
[17] 夏贯芳,刘廷禹,张涵,等.硅酸镥晶体本征缺陷和力学性质的研究[J].人工晶体学报,2013,42(12):2691-2695.
[18] WILLIFORD R E,DEVANATHAN R,WEBER W J.Computer simulation of displacement energies for several ceramic materials[J].Nuclear Instruments & Methods in Physics Research,1998,141:94-98.
[19] 李妍,刘廷禹,赵朝珍.磷酸镓晶体物理性质及本征点缺陷的研究[J].人工晶体学报,2012,41(5):1366-1370.
[20] 马昌敏,刘廷禹,常秋香,等.ZnO和ZnS本征点缺陷的理论研究[J].高等学校化学学报,2016,37(5):932-937.
