二氧化碳电化学还原的研究进展
2018-01-15白晓芳王白银冯光辉孙予罕
白晓芳 陈 为 王白银 冯光辉 魏 伟 焦 正 孙予罕
二氧化碳电化学还原的研究进展
白晓芳1,2陈 为2,*王白银1,2冯光辉1,2魏 伟2焦 正1孙予罕2,*
(1上海大学环境与化学工程学院,上海200444;2中国科学院低碳转化科学与工程重点实验室,中国科学院上海高等研究院,上海201210)
利用低品阶的可再生电能,将二氧化碳 (CO2)电化学还原生成高附加值的化学品或燃料,既可以“变废为宝”、减少 CO2排放,又能将可再生能源转变为高能量密度的燃料储存,具有重要的现实意义。电化学还原CO2的研究,是目前世界范围内的研究热点,许多标志性的重要研究成果不断涌现。本文首先简要介绍了 CO2电化学还原的基本原理,然后概述了近 5年来在其电催化剂材料和反应机理相关的实验与理论研究方面的昀新研究进展,昀后对其发展趋势进行了展望。
二氧化碳;可再生能源;电化学还原;电催化剂;反应机理
1 引言
现代社会消耗了大量的化石能源(煤、石油、天然气),产生了大量二氧化碳(CO2)进入大气,由此引起了温室效应等一系列的全球气候环境问题1。2016年4月联合国大会通过的《巴黎协定》提出了2020年后全球应对气候变化、实现绿色低碳发展的蓝图和愿景,以实现在2100年之前将全球平均气温较工业化前水平的升高幅度控制在2 °C 范围之内。
CO2作为一种经济的、安全的、可持续的碳氧资源化合物,将其转化为液体燃料、化学品的发展潜力巨大。但是由于CO2化学性质非常稳定,需要施加额外的能量才能使其活化、转化。在实际工业过程中能够利用CO2的反应不多,例如,尿素合成、碳酸酯合成、甲醇合成、甲烷的CO2重整等2–5。这些化工过程,一般需要高温、高压等较为苛刻的反应条件,是高能耗、低效率的过程。另一方面,近年来,我国在新兴能源领域发展迅速,2016年可再生清洁能源发电(风电以及太阳能发电)高达22606万千瓦,占装机总量的近14%6。但风能、太阳能等具有很强的随机性、间歇性、波动性及反调峰性等特点,对电网的冲击较大而无法并网,造成了这些可再生能源的较大浪费。
从资源、能源发展战略的角度来看,利用低品阶的可再生电能将CO2高效电化学还原成化学品或燃料,既可以“变废为宝”、减少CO2排放,又能减轻人类对化石燃料的依赖,对于缓解能源与环境双重压力具有重要的现实意义。
近年来,电化学还原CO2的研究在世界范围内掀起了热潮,大量的重要成果不断涌现。本文在简要介绍电化学还原CO2的基本原理的基础上,着重对最近5年内CO2电化学还原领域的最新研究成果进行了较详细的归纳、总结,从不同类别的催化剂研究进展、反应机理的研究方法与手段等方面进行了分析、讨论,并对其发展趋势进行了展望。

2 电化学还原CO2的反应原理
CO2在环境条件下是非常稳定的,其还原反应需较高的负电势来驱动。同时,有几个质子伴随着多电子的转移过程也需要相似的电势,这导致产物选择性控制变得非常困难。例如,还原反应可通过2、6、8和12电子转移过程(表1)7和反应产生一系列的产物,包括一氧化碳(CO)、甲酸(HCOOH)、甲醇(CH3OH)、甲醛(HCHO)、甲烷(CH4)、乙烯(CH2CH2)、乙醇(CH3CH2OH)、乙酸盐(CH3COO−)和其他产物8。此外,在水溶液中进行CO2电催化还原通常都伴有析氢反应(HER)发生。因此,高活性的CO2还原电催化剂在反应中不仅要有高效率和高选择性还要能抑制氢气的产生。
现有的CO2还原电催化剂可以分为三类:金属催化剂,非金属催化剂和分子催化剂9–24。早期CO2电化学还原的研究主要集中在多晶单金属催化剂上,因为它们结构简单,易于处理,所以它们成为基础研究的首选。基于CO2还原的主要产物,单金属催化剂可以进一步分成几个亚组:CO选择性金属(如Au、Ag 和Zn),甲酸盐选择性金属(如Sn、In、和Pb)和氢气选择性金属(如Fe、Ni和Pt)等。在所有单金属催化剂中,Cu表现出独特的电催化能力,可生成甲烷、乙烷、乙烯、乙醇等小分子烃类及醇类等产物25。目前的CO2电催化剂已经不再局限于体相单金属催化剂,而诸如纳米结构金属10–12,离子改性金属13–15,26,双金属16,17,27和非金属材料20,21,28等新型催化剂也被证明是非常有前景的电化学还原CO2材料,它们代表了CO2电催化还原的新趋势。
3 电化学还原CO2的催化剂
传统的CO2电催化的主要研究对象是体相的贵金属催化剂。但是它们在不改变催化剂材料的情况下,已不能改善反应活性和反应速率。因此,越来越多的研究者趋于新型催化活性位点或催化剂结构的研究,以提高电化学CO2还原过程的效率。目前已经开发出新的催化剂类型包括以下几种,例如金属纳米材料,金属氧化衍生物29,有机无机杂化材料30等。
3.1 金属催化剂
具有纳米结构的金属催化剂材料,一般具有高的比表面积,表面活性位点的数目也随比表面积成比例增加,因此纳米金属催化剂会有更多的活性位点31。更重要的是,纳米结构金属催化剂表面含有大量的边缘/低配位点,这可能使其表现出不同于体相金属的行为。纳米催化剂颗粒的几何形貌,粗糙度和尺寸可能对CO2 还原过程中的活性和产物选择性产生影响32–34。新型金属催化剂类型已被开发和应用,如纳米材料(如纳米粒子,纳米管,纳米线)10–12,35,36,纳米多孔膜11,12等。
Lu等11在HCl 溶液中,以Ag-Al 为前驱体,通过两步去合金法制备了纳米多孔银催化剂(np-Ag)。通过选择性蚀刻Al,剩余的Ag 原子重组、形成了三维的纳米多孔结构。图1(a–c)显示了np-Ag 的形态、结构。当过电位小于0.5 V 时,np-Ag催化剂的CO2还原生成CO的选择性为92%,其反应速率是多晶Ag 的3000倍。具有较高的催化活性可能是因为CO2−中间体在高度弯曲表面上的稳定性更高,所以克服热力学阻碍所需的超电势较小。这与图1d中np-Ag催化剂具有较小的塔菲尔斜率的结果相一致。np-Ag 的电化学表面积是多晶Ag的150倍左右,np-Ag的CO2还原活性是多晶Ag的20倍。

表1 CO2电化学还原的半反应及对应的电极电位7
Hsieh等15在AgCl溶液中使用氧化还原方法制备了珊瑚状的纳米结构的Ag电催化剂。他们发现Cl−与Ag纳米珊瑚表面形成了稳定的化学键。这种材料与多晶Ag相比,其电化学表面积增加了20倍,具有比较高的催化活性:Cl−键合的Ag纳米珊瑚在还原CO2时,CO的分电流密度增加了660倍,催化活性提高了32倍。作者认为Cl−的存在不仅有利于制备高比表面积的的纳米Ag催化剂,还能提高CO2还原的活性,并抑制氢气的生成,从而提高CO的选择性。
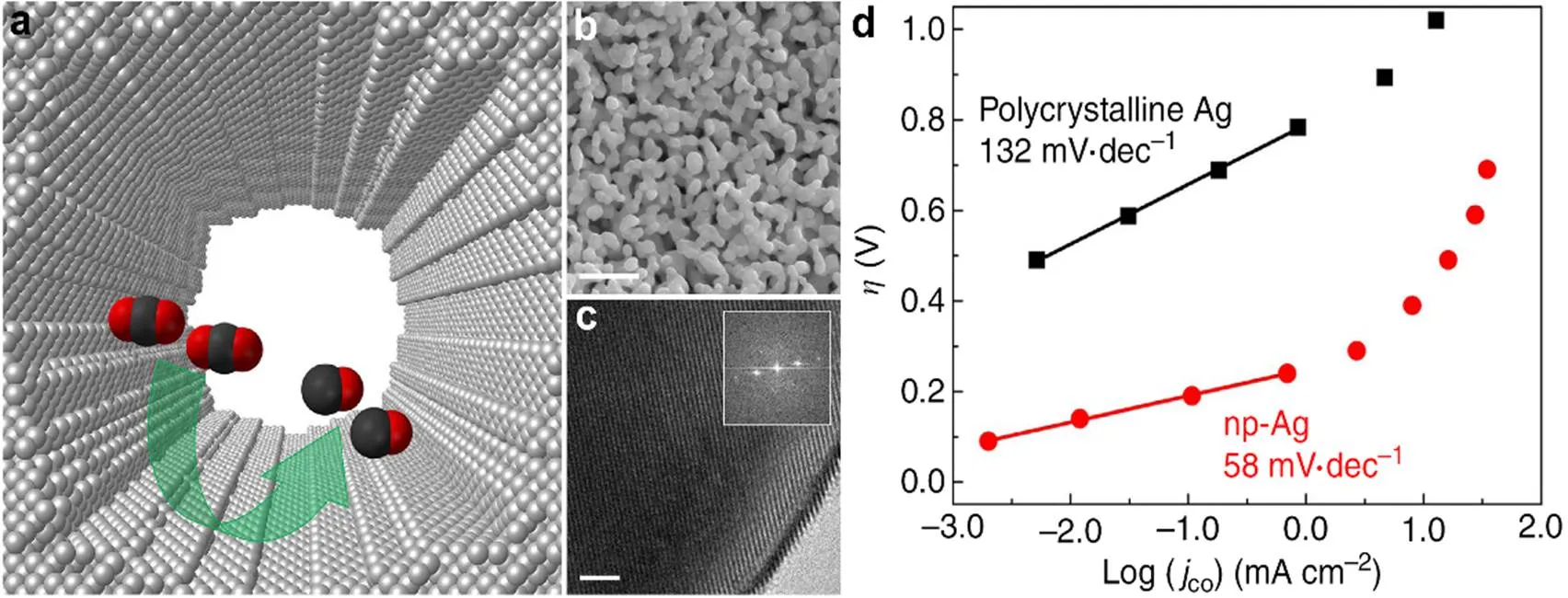
图1 np-Ag的表征和塔菲尔分析11
(a) A schematic diagram of a nanopore of the silver electrocatalyst with highly curved internal surface. (b) Scanning electron micrograph(scale bar, 500 nm). (c) High-resolution transmission electron micrograph (scale bar, 2 nm). (d) Tafel slope analyses.
不同的纳米尺寸结构对CO2电催化性能也有一定的影响37,38。Zhu 等39对不同纳米尺寸的Au纳米颗粒在0.5 mol∙L−1KHCO3电解质溶液中进行了CO2电还原研究。颗粒的纳米尺寸为4、6、8和10 nm,其中粒径为8 nm的Au纳米颗粒的催化效果最好,在−0.67 V (RHE)下,总的法拉第电流效率达到90%。密度泛函数理论表明,边缘位点多有利于CO的生成,而角位点多有利于H2的生成,Au纳米颗粒表面边缘位点较多,因此有利于中间产物COOH*和CO的生成。8 nm的金纳米颗粒的边缘位点与角位点的比值最大,因此具有最好的选择性(图2)。

图2 金纳米粒子的表征和电化学性能39
TEM images of (a) the 8 nm Au NPs and (b) the C-Au NPs. (c) CO FEs. (d) Current densities for CO formation.
Gao等40制备了不同纳米尺寸(2.4–10.3 nm)的Pd纳米粒子进行CO2还原,发现在−0.89 V (RHE)电位下,3.7 nm的Pd纳米颗粒的CO法拉第电流效率为91.2%,而10.3 nm的只有5.8%,3.7 nm的Pd纳米颗粒是10.3 nm的18.4倍(图3)。

图3 不同尺寸的Pd纳米粒子表征和电催化性能40
TEM image and HRTEM images of (a) 3.7, (b) 6.2, and (c) 10.3 nm Pd, (d) Faradaic efficiencies and (e) current densities for CO production.
合金化也是一种提高CO2还原的催化活性和选择性的重要方法41,42。通过合金化,可调节中间体在催化剂表面上的结合强度从而提高CO2 还原反应动力学。动力学中反应的难易主要与反应的能垒有关。催化剂表面上的活性位点与中间体的结合能力越强,则中间体越稳定,反应的能垒越低,越有利于反应的发生。Kim等16研究了通过金属前体的共还原合成的双金属纳米颗粒Au-Cu合金催化剂,使用前体比来控制合金组成。电化学测试表明,甲烷和乙烯的法拉第效率随着铜含量增加而增加,但是CO法拉第效率呈现出了火山型的变化趋势。在−0.73 V (RHE)电位下,Au3Cu在所有研究的组合物中表现出最高的周转率(TOR)和CO的部分电流密度。这表明双金属AuCu组合物的活性/选择性是几何效应和电子效应协同作用的结果。
Luc等43使用Ag-Sn双金属芯和超薄的部分氧化的SnO壳,制备了一系列不同Ag/Sn比例的具有核-壳结构的纳米Ag-Sn电催化剂。密度泛函理论(DFT)表明,SnO和它的氧空位是CO2还原的重要活性组分。Ag-Sn核壳结构中Sn的浓度越高,中间体OCHO*越稳定,越有利于产物甲酸的生成。在1.7 nm的最佳SnO壳厚度下和电位−0.8 V下,催化剂具有约80%的甲酸法拉第效率和16 mA·cm−2的甲酸盐分电流密度。
Zhong等44使用新型电化学沉积法直接在处理过的碳纸上生长纳米结构的树突状铋催化剂。它具有优异的CO2还原性能,甲酸的最大法拉第效率可达96.4%,电流密度为15.2 mA·cm−2。而且催化剂可以连续稳定电解10 h。Lv等45在Cu箔上沉积了一种新型纳米尺寸的Bi基催化剂,此Bi/Cu电极可以在较低的超电势下(0.69 V (Ag/AgCl))将CO2还原为甲酸,在沉积时间为25 min下制备的Bi/Cu电极可得到最大的甲酸盐法拉第效率(91.3%)。
3.2 金属氧化物催化剂
最近,金属氧化物衍生纳米结构催化剂已经被报道可以在低电位下进行CO2高选择性还原29,46–48。然而,目前尚不清楚这些氧化衍生金属催化剂的电催化活性是如何受到它们的氧化物影响的,可能是因为它们的微结构特征,如界面和缺陷49等影响着CO2还原路径。
Gao等50先将乙酰丙酮钴Co(acac)3水解成[Co(H2O)6]3+,然后冷凝生成片状的产物。水解过程中加入正丁胺以降低表面能,避免聚集。接着在220 °C反应3 h生成纯的Co原子层,反应48 h生成部分氧化的Co原子层(图4)。通过TEM、原子力显微镜等表征手段得出两种物质都是四个原子层厚度。在0.1 mol∙L−1Na2SO4溶液中和−0.85 V (SCE)电位下,部分氧化的四层原子厚度的Co催化剂的电流密度为10.59 mA∙cm−2,是纯四层原子厚度的Co、部分氧化体相Co以及体相Co的10、40和260倍。原子层的部分氧化进一步增强其内在活性,实现了超过4 h的稳定电流密度(约为10 mA∙cm−2),大约90%的甲酸盐选择性,而且只有0.24 V的过电位,超过以前报告的金属或金属氧化物电极29,47,51–53。正确的形态和氧化态可以将一种被认为几乎没有催化活性的材料转变为CO2电还原反应活性催化剂。Gao等54进一步研究了Co3O4单元层结构中的O(II)空位对CO2电催化的影响。他们用十六烷基三甲基溴化铵(CTAB)和乙酰丙酮钴Co(acac)3自组装生成Co(CO3)0.5(OH)∙0.11H2O-CTAB 层状杂化物,再自剥成Co(CO3)0.5(OH)∙0.11H2O原子层,氧气条件下320 °C煅烧5 min生成少量氧空位的Co3O4单元层,而在空气中则生成富氧空位的Co3O4单元层。他们发现O(II)空位有助于CO2吸附,通过DFT计算发现O(II)空位的存在可以提高限速步骤HCO3−的质子脱离速度,从而有助于HCOO−生成。O(II)空位的存在通过稳定甲酸根阴离子基中间体使速率限制活化能从0.51降低到0.40 eV,起始电位从0.81降低到0.78 V,塔菲尔斜率从48 降低到37 mV·dec−1。因此,富氧空位的Co3O4单元层在0.87 V 电位下稳定运行40 h,获得2.7 mA·cm−2的电流密度,甲酸的法拉第电流效率达到了85%。

图4 220 °C煅烧3h得到的部分氧化4原子层厚度Co的表征50
(a) High-resolution TEM image. (b, c) Enlarged high-resolution TEM images. (d, e) The related schematic atomic models. (F–h) Elemental mapping. (i) Faradaic efficiency of formate.
Li等29在空气中对Cu箔进行煅烧。通过控制煅烧的温度和反应时间来控制Cu2O层厚度。−0.5 V (RHE)电位下对多晶铜和不同Cu2O层厚度的电极进行CO2还原,结果表明500 °C煅烧12 h得到具有较厚Cu2O层的Cu电极具有较好的还原效果,其中反应稳定后总的电流密度达到2.7 mA·cm−2,CO和HCOOH 的法拉第电流效率分别为40%和33%。反应后的SEM图如图5a所示。图5b比较了多晶铜和500 °C煅烧12 h得到具有较厚Cu2O层的Cu电极在不同电位下的还原产物,发现有Cu2O层可以在较低的反应电位下生成较多的CO和HCOOH。Cu电极的活性与Cu2O层厚度有关:需要大于1 μm的Cu2O层才能大大提高催化剂的CO2还原活性。
Mistry等55在O2和H2等离子体下电解抛光得到氧化铜催化剂(图6)。通过控制等离子体的条件,可以控制Cu的表面和氧化物的形态和厚度。这种氧化铜催化剂可以在较低超电势下电还原CO2,并且在−0.9 V (RHE)电势时生成乙烯(60%)的选择性最高。另外,采用扫描透射电子显微镜-能谱(STEM-EDS)和X射线吸收精细结构谱(XAFS)的方法,发现了表面的氧化物层在−0.91 V(RHE)反应1 h后,仍能保持较好的稳定性。催化剂表面主要是Cu+,所以粗糙的铜氧化衍生物仅起部分作用,而Cu+的存在才是降低电位和提高乙烯选择性的关键。

图5 (a)多晶Cu箔500 °C煅烧12 h在−0.5 V vs RHE电位下进行CO2还原反应后的SEM图和(b)产物CO和HCOOH 的法拉第电流效率29

图6 等离子体激活铜箔的形态和化学分析55
EDS elemental maps of Cu foils treated with O2plasma for before and after reaction, (a) 20 W 2 min;(b) 100 W 2 min; and (c) 100 W 2 min + H2plasma. (d) Faradaic efficiency of C2H4.
Au的金属氧化物层的厚度对CO2还原活性具有一定的影响。恒定电位阳极氧化对形成非常薄的氧化物层很有效,而施加阴极电位后几秒钟内氧化层就会被还原。Chen 等46在0.5 mol∙L−1H2SO4中施加1 kHz的周期性对称方波电位程序,制备大于1 μm的非晶Au 氧化层,所得电极在NaHCO3电解质中进行CO2还原。其CO选择性非常高,而且超电势低至140 mV,并能反应8 h不失去活性。
Zhang等51报道了具有受控粒径的超细纳米氧化锡上还原CO2的电化学研究结果。通过水热法将氧化锡纳米粒子负载在具有高比表面积的碳载体上,利用它们的3D多孔结构以促进CO2的运输和还原。控制碳载体上的SnO2的纳米颗粒尺寸,CO2还原生成HCOO−的过电位可以低至340 mV。以高比表面积的石墨烯为载体时甲酸的最大的法拉第效率高达93%,同时电流密度为10 mA∙cm−2。
3.3 有机无机杂化催化剂
金属络合物催化剂(如过渡金属复合物)可以通过系统的改变化学结构进而提高反应活性和选择性;特别是它们的纳米结构组装可以提高它们的活性位点数量和加快反应速率56–59。最近报道,具有可调控的分子单元的共价有机骨架(COFs)和金属有机骨架(MOFs)催化剂可以提高CO2还原的活性和选择性。Lin等60,61研究发现带有Co卟啉的COFs将CO2电还原为CO的反应具有高活性和高选择性。通过亚胺键连接钴卟啉催化剂与有机骨架制备得到多孔COFs材料(图7)60。通过增加有机骨架长度和用铜稀释钴催化剂,使得COFs中的孔径增大,其暴露于反应物的活性位点比例增加,导致每个钴单元的周转频率增大。这种有机骨架催化剂在过电位−0.55 V下的法拉第电流效率高达90%,周转数大于290000,是分子钴复合物催化剂活性的26倍。X射线吸收光谱(XAS)用于评估周围的COF如何影响钴卟啉单元的电子结构。在CO2气氛中,对COF催化剂施加一个还原电位(−0.67 V (RHE)),Co的边缘XAS谱图中线性变化趋势与部分Co(II)还原成Co(I)是一致的。该催化剂具有钴卟啉分子所不具有的前边缘特征,这表明COF的环境可以直接调节分子中心的电子结构,并将其耦合延伸至外晶格。金属和COF晶格的相互作用力给钴中心提供更多的非定域化的电子结构。除了COF,钴卟啉也可以复合MOF中,如Kornienko等62将Al2(OH)2TCPP-Co (TCPP = 1,4′,4″,4‴-(porphyrin-5,10,15,20-tetrayl) tetrabenzoate)复合到MOF中。合理选择活性位点、无机框架、厚度/负载量,将所制备的MOF集成到导电载体上形成一层薄膜。这种MOF催化剂电催化CO2生成CO的选择性高达76%,稳定性超过7 h,每个活性位点的TON为1400。原位光谱电化学测量证明了Co(II)的活性中心通过MOF催化剂被还原为Co(I),然后开始还原CO2。另外,Shen等30将钴原卟啉固定在热解石墨电极上可以在中等超电位(0.5 V)的酸性水溶液中进行CO2还原生成CO和其它较少的产物(例如甲烷)。基于实验结果,提出了初始中等CO2自由基物种作为一个强大的强碱基与催化剂表面结合从水分子提取质子的理论30。Co原卟啉反应机理示意图如图8所示。

图7 基于钴卟啉的2D共价有机骨架的填充空间结构模型和电化学性能60
(a) structural models, (b) Cyclic voltammograms, (c) Tafel plots.

图8 原卟啉上电化学还原CO2法拉第效率和反应机理示意图30
(a) Proposed mechanistic scheme, (b) FE of CH4, (c) FE of CO.
同时,Hod等63将Fe-卟啉和MOF-525复合用于CO2还原。发现Fe-MOF-525薄膜中Fe-卟啉表面浓度高(1015 sites∙cm−2),这相当于900单层表面吸附的Fe-卟啉。实验结果证实了用COF或MOF获得的多孔结构分子催化剂用于CO2还原的可行性。
除了上述杂化分子材料体系外,一些基于Cu和Zn的MOF也可以直接用作电催化剂用于CO2还原。Hinogami等64应用具有电子传导性,质子传导性和均匀分散的反应位点的特点的铜硫酸盐MOF作为催化剂进行CO2还原。有趣的是,铜硼酸盐MOF在水溶液电解质中−1.2 V (SHE)电位下只有HCOOH产物生成,而Cu金属电极则会产生了一系列的产物。另外,HCOOH的产量是Cu金属电极的13倍。后来,Kumar等65证明了Cu3(BTC)2(BTC = 1,3,5-苯三甲酸)MOF膜在含有四丁基–四氟硼酸钡的N,N-二甲基甲酰胺中是一种选择性还原CO2生成草酸的有效的电催化剂。最近,Kang等66在碳纸上沉积Zn-1,3,5-苯三甲酸金属有机骨架(Zn-BTC MOF)在以离子液体为电解质的体系中作为电催化剂进行CO2还原。Zn-MOF的形态对性能有显著的影响,片状Zn-MOF由于其较大的电活性表面积而表现出最高的催化活性。通过比较不同的离子液体,发现含氟的咪唑鎓离子液体与CO2的相互作用最强。Zn-MOF和离子液体的优异组合导致在0.25 V的超电势下,CO2转化为CH4的电流效率大于80%,电流密度大于3 mA∙cm−2。MOF基催化剂和离子液体的组合为提高CO2碳还原效率提供了一个替代系统。尽管大多数MOF和COF具有极限的化学稳定性,但是这可以通过不同的方法来改善,例如利用高价态金属离子或氮供体配体形成的强金属-氮键,或引入多孔聚合物结构(包括稳定的共价键,C―C,C―H和C―N键)等。
3.4 其他类型催化剂
铜是一类特殊的金属,它可以电催化CO2生成乙醇、乙烯等C2产物67,68。虽然Cu比其他可以电催化生成CO的电极比更便宜、储量更丰富,但是活性差,为了让CO和甲酸的选择性更高要避免使用多晶铜电极69。无机中空纤维和微管电极已被用于固体氧化物燃料电池。这种电池制备成本低,而且可以高功率输出70。含水电解质,由镍和碳组成的中空纤维也被用作阴极用于氢或氧还原71,72。Kas等73制备了三维多孔中控纤维铜电极。三维多孔中控纤维铜作为气体扩散器和阴极可以产生非常高的CO,甚至与使用贵金属相当。在CO2流速为30 mL∙min−1的流速和电位0.4 V (RHE)下,CO的法拉第电流效率达到75%。这主要归因于其丰富的多孔结构。
对于一些化学活性强的金属,如果在表面暴露大量的原子,它们在环境条件下非常不稳定,容易被氧化29,47,48,51,这将降低电子导电性,从而导致催化剂活性快速衰减。将金属超薄层嵌入到石墨烯层间结构中,金属超薄层不仅可以作为“间隔”有利于电解质扩散到石墨烯的基质中74,而且可提供丰富的表面原子作为活性位点有利于CO2吸附75,76。高导电性石墨烯可以使电子快速穿透到金属超薄层77。Lei等78将金属Sn量子片限制在石墨烯中发现其具有更高的CO2电催化活性(图9)。限制在石墨烯中的Sn量子片在−1.8 V (SCE)电位下的电流密度是21.2 mA∙cm−2,分别是15 nm的Sn纳米颗粒与石墨烯、15 nm Sn纳米颗粒和体相Sn混合的2、2.5、13倍。而且它可以长时间稳定运行长达50 h,并且甲酸的法拉第电流效率达到85%。
另外,在碳纳米结构中的缺陷中引入杂原子也可以改善CO2催化活性79–83。在金属催化剂中只有Cu催化剂才能生成C2产物,而Wu等84用一种无金属纳米尺寸的N掺杂石墨烯量子点的电催化剂进行CO2电催化还原也可以产生C2产物。这种N掺杂石墨烯量子点的CO2还原总法拉第电流效率可达90%,其中乙烯和乙醇的总法拉第电流效率达到45%。这对于电催化CO2转化生成C2产物的研究具有参考意义。

图9 限制在石墨烯中的Sn量子片的形成过程和表征78
(a) Scheme illustration for the formation of Sn quantum sheets confined in graphene, (b) TEM image, (c) HRTEM image, (d) AFM image, (e) electrocatalytic performances of the composites at different potentials.xx
4 电化学还原CO2过程的机理
对于CO2的电化学还原反应,发展高活性、高选择性和高稳定性的电催化剂及其催化体系的一个主要障碍是缺少对电化学反应过程机理的深入理解,从而无法从本质上设计、构建催化剂。下面我们分别从反应动力学、原位表征和DFT计算三个方面来介绍在电化学还原CO2反应机理的研究进展。
4.1 反应动力学
动力学现象研究已经被应用于电化学CO2还原12,46,例如,Prakash等85比较了三种锡电极在NaHCO3水溶液中的CO2还原反应。三种锡电极分别为:Sn粉修饰的气体扩散层电极(SnGDL)、锡金属圆盘电极(SnB)和Sn粉末涂层的石墨电极(SnG)。作过电位()相对于电流密度()的曲线,可以发现该曲线的线性区域服从于塔菲尔方程。在该类型电极上CO2反应的交换电流密度(A∙cm−2)表明相对于SnG电极,SnGDL电极相对增加了五倍左右,该密度由电流-电压曲线的塔菲尔图确定。而且,SnG电极的交换电流密度比SnB电极高出两个数量级。电解中,在电压为−1.6 V(NHE)时,电流密度达到最大,其最大值是27 mA∙cm−2,此时甲酸盐的法拉第效率为70%,这可能是迄今为止文献报道的在环境压力下,Sn电极上法拉第效率的最高值之一。
改革开放40年中国社会经济的发展,是中国设计走向体系化、市场化,释放自身能量的过程。然而,回忆过往,中国能够建立全面、完整的工程设计体系,那些工程设计院(所)功不可没。
Kim等86报道了Mo2C在低电势下还原CO2生成甲烷的能力。相对于Cu电极,随着外加电势从−0.8降到−1.1 V时,其生成甲烷的法拉第效率从0.03%增加29%,而且也会有少量的C2H4和C3H6生成。由甲烷的分电流密度的Tafel曲线可以看出,只有一部分的电流可以归因于甲烷的生成。其中,Cu电极的Tafel斜率为−54 mV∙dec–1,而Mo2C粉末的Tafel斜率为−297 mV∙dec–1。这也就是说,Mo2C比Cu的甲烷动力学具有更弱的电势依赖性。Tafel斜率计算点的数据的电势低于−1.0 V(RHE),这样避免了斜率在更负电位下的剧烈变化。这些结果意味着在Mo2C电极上的甲烷形成机理与在Cu电极上明显不同,但Mo2C电极的制备方法是相对独立的,且生成甲烷的更陡的Tafel斜率,表明动力学相对于外加电势只有微弱的函数关系。
Hatsukade等87发现还原产物的分电流密度是由每个电势下的总电流密度乘以当前每个产品的效率而得到的,塔菲尔斜率图如图10所示。这些数据常常有助于CO2还原反应的动力学和机理研究,因为局部电流密度与周转频率(TOF)成正比87。为了方便讨论,作者在以下三种不同的电势区域内进行讨论:低超电势(−0.6 – −1.0 V (RHE))、中超电势(−1.0 – −1.2 VRHE)和高超电势(−1.2 – −1.4 VRHE)。值得注意的是,文中定义的低超电势区域是相对的,即是相对于CO2还原反应计算出的标准还原电势,高于0.5 V的超电势区域。如图10所示,在低超电势区域只观察到三种还原产物:H2、CO和甲酸盐,其中每种产物的生成需要2个电子。CO的TOF随着超电势作用的增加而增加,塔菲尔斜率为140 mV∙dec−1,然而H2的TOF在该电势区域保持恒定。这表明在该电势区域,产物的选择性从H2向CO转变88。在中超电势区域,CO的TOF突然到达最高值,这也导致总电流密度出现一个短暂的高峰,因为CO是该电势区域的主导产物。也值得注意的是该电势区域中H2的TOF仍然保持恒定,且甲酸盐的TOF随着超电势的增加而增加,但是相对于H2和CO,甲酸盐的TOF依然保持在低水平。在高超电势区域,可以观察到CO的TOF降低,而H2的TOF增加,这说明在该电势区域的产物选择性向H2转移。在该区域产物的形成需要多于2个电子,且他们的TOF随着超电势的增加而增加。

图10 (a)不同电位的电流密度,(b)产物的分电流密度的塔菲尔曲线87
Lv等45合成了一种新型沉积在铜箔上的纳米级铋基催化剂,该催化剂在低过电位下可以高选择性的电化学还原CO2生成甲酸盐。人们普遍认为,在热力学上,还原CO2生成CO2•−发生在−2.1 V (Ag/AgCl)电极。机理主要依赖于对Tafel斜率的解释,作者对Bi/Cu电极的Tafel斜率进行了研究。根据所得的塔菲尔曲线,Bi/Cu电极的Tafel斜率为128 mV∙dec−1,这与118 mV∙dec−1很接近,而后者的Tafel斜率表明该反应通过CE机制发生,其中CE机制指的是CO2和碳酸盐之间的化学平衡之后是限速电子转移的过程。
由于动力学研究是用来探测CO2电化学还原反应机制的一种有效的手段,因此提出一种定义明确的动力学研究方法是非常棘手的。例如,尽管在含有电子转移的反应中,塔菲尔分析常常被用来确定反应的速度控制步骤或主要反应中间体的表面覆盖度,然而其斜率是基于许多因素形成的。总之,对动力学的研究来说,产物或反应物浓度相对于标准氛围的偏差,可能会导致从热力学预测值得到的平衡电势的改变。
4.2 原位表征
Baruch等89使用原位衰减全反射红外光谱(ATR-IR)来对Sn电极界面的CO2还原反应进行实时检测。原位光谱证据表明在CO2还原的过程中,表面有氧化锡的存在。在ATR-IR谱中,2350 cm−1处的双重峰对应于气相的CO2,1470和1420 cm−1处的强吸收峰表明游离的碳酸根离子的存在,单配位锡碳酸盐在1530−1470 cm−1和1370−1300 cm−1存在两个强吸收峰,这证明了表面结合锡碳酸盐的存在,为一个重要的活性中间体。在CO2还原反应中,原位衰减全反射红外光谱表明在Sn/SnO薄膜上确实存在碳酸盐吸收,且表面氧化物和碳酸盐的作用也被阐明。这与Baruch等和Qiao等采用的塔菲尔斜率研究相关联34,89,提出了一种CO2还原生成甲酸的机制,该机制充分考虑了作为一个重要的反应中间体的表面结合碳酸盐的种类,并且否定了CO2•−自由基的生成。Fang等90比较了新鲜制备的MPA-Au相同样品(MPA:2-巯基丙酸)在−0.94 V (RHE)电解后和在−1.00 V (RHE)电解后的ATR-IR光谱(图11)。在新制备的电极光谱中,1723 cm−1(a)处峰对应于COOH基团的C=O伸缩振动,―CH3基团的对称和不对称伸缩振动分别在1372 cm−1(b)和1449 cm−1(c);C―C伸缩振动在1241 cm−1(d)70,91;1607 cm−1处轻微肩峰(e峰)和1421 cm−1处小峰(g峰)分别对应于―COO−的不对称和对称拉伸。在电解后(−0.94 V (RHE))的MPA改性Au样品的光谱上,1583 cm−1(e)处的不对称伸缩振动和1662 cm−1(f)处的肩峰,分别表明了去质子化的COO−基团和COOH基团的存在。峰(g)与峰(e)的上升表明,去质子化的配体物质在电解之后占主导地位92。在电解前和电解后的样品中,CH3基团和C―C键的振动保持相似;波数的微小变化表明,单层配置的变化导致Au表面和官能团之间更强的相互作用。然而,在更负的电位下(−1.00 V (RHE)),与―CH3基团和COO−基团相关的吸光度的降低,表明了表面配体浓度的降低,这说明在低于−0.94 V (RHE)时,MPA保持在表面。
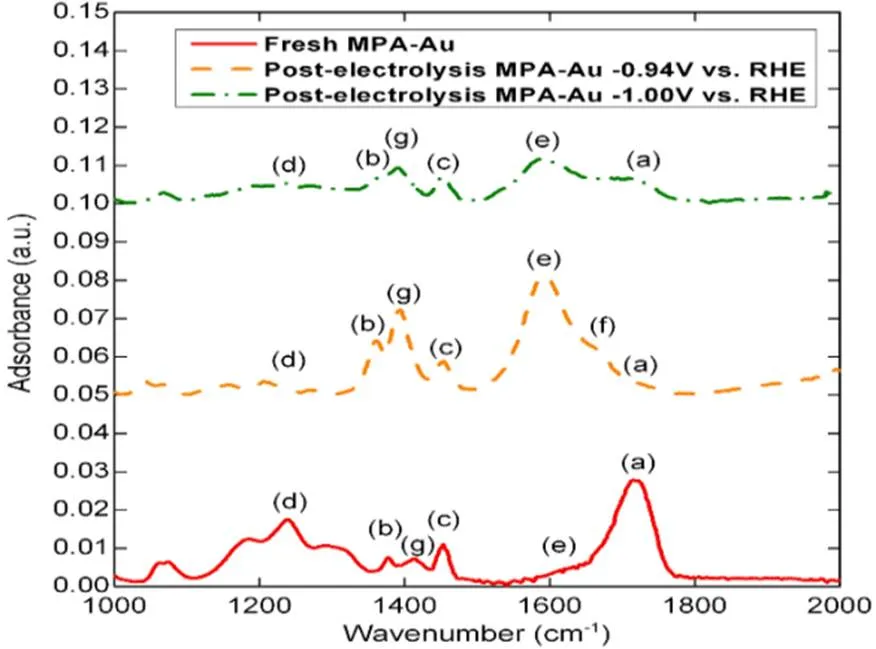
图11 MPA修饰的Au的ATR-IR光谱的比较90

图12 过渡金属和分子催化剂上的CO2电催化还原生成产物的可能反应途径7
(a) pathways from CO2to CO, CH4, CH3OH, and HCOO–; (b) pathways from CO2to ethylene and ethanol; (c) pathway of CO2insertion into a metal–H bond yielding formate.
4.3 DFT理论计算
多重质子电子的热力学理论只适用于以下情况:2电子转移过程、氧化还原反应且反应过程中过电位是可忽略的反应。这种情况被称为“可逆的”催化。例如,在铂电极上的氢氧化和析氢反应就是很好的可逆催化的例子。值得注意的是,可逆催化剂满足Sabatier规则,这意味着它们的反应中间体以最佳的方式存在。在这种情况下最好的状态是结合力适中;过强的结合将导致催化剂中毒,同时太弱则不能开始反应。对于2电子转移反应,通常只有一个中间体,因此,最佳的催化剂是“简单”一维优化理想催化剂。现在通过量子化学计算第一原理可以通过推测催化中间体准确计算结合能,通常采用密度泛函理论(DFT)94。Kortlever等7用DFT计算,对过渡金属和分子催化剂上的CO2电催化还原生成不同产物的可能反应途径进行研究,如图12所示。图12a,研究的是电还原CO2生成CO、CH4、CH3OH、HCOO−的途径。图12b,是CO2还原成乙烯和乙醇的途径。图12c,CO2在金属–H键中反应生成甲酸盐的途径。通过该研究,可以进一步的了解了CO2还原的反应过程,为设计、制备高活性、高选择性的催化剂提供参考。
5 展望
虽然CO2电催化还原技术不断的发展,但是尚未达到商业化的要求。理想的电催化剂应该达到以下几个要求:高活性,低电位,对所生产产物具有高选择性,可以长期稳定的运行,更重要的是,是地球丰富元素的组合物,而不是局限于几种稀缺的贵金属。为了实现这些目标,需要从以下几个方面进行努力:
(1) 通过引入多级孔结构探索新型非贵金属电催化剂
在许多电化学反应中多孔材料具有非常突出的催化性能,如氧还原反应和析氢反应95–98。从微孔到大孔区域的多孔材料的孔径大小可以调节电解质的扩散。孔径大于50 nm的是大孔,它有助于反应过程中的反应物和产物在电极之间的扩散和转移,而介孔(2–50 nm)和微孔(< 2 nm)却拥有大的表面积,从而生成更多的高分散的活性位点。因此,具有多级孔结构的材料可以加速物质的运输和提高CO2电催化转化的效率。例如,去合金法得到的多孔Ag催化剂具有高的比表面(是未处理的Ag比表面积的150倍),和大量的高活性位点(至少是未处理的Ag的20倍)11。更有意义的是发现通过胶体晶体模板法制备的介孔的Au催化剂可以调节CO2还原催化选择性99。介孔结构可以控制扩散反应,抑制氢气的产生和保持CO2电还原生成CO的高速率。因此,对于非贵金属催化剂提升其电催化CO2还原性能,通过引入多级孔结构是非常有效的方式。
(2) 通过均相和非均相系统优化分子水平的活性位点
分子催化剂(如吡啶盐和金属卟啉)在CO2电还原领域存在着两大挑战:减小过电位和增大所需产物的选择性100。然而,均相催化体系反应只在导电电极上以及电极附近才能进行,因此很难获得高的电流密度。此外,在均相溶液体系中分子催化剂的溶解度也是一个备受关注的难点。反之,非均相电催化系统则可以克服这些障碍。在非均相系统中,催化剂不容易失活,催化剂活性累加,可以更好的控制化学环境下的活性位点63。具有固体支撑的分子催化剂相对于没有支撑的分子催化剂来说会有更高的电流密度。钴卟啉修饰的COFs和MOFs的良好的性能证明了这一概念的合理性。如果分子催化剂的选择性和催化能力可以与固体多相催化剂的特点相结合,那么阻碍CO2电化学还原的主要问题可能得以解决。
(3) 通过原位表征技术和理论计算了解CO2电还原机制
对CO2在不同电解质(特别是水溶液体系)中的还原机制的了解是发展高效的电催化剂的先决条件。各种原位表征技术可以直接探测还原反应过程,并获得反应机理,它可以用来推测高效催化剂的结构和组成。例如,使用原位衰减全反射红外光谱可以直接观察到在Sn催化剂表面和碳酸盐介质的中间活性物质89。用原位紫外可见吸收光谱观察钴卟啉修饰MOFs催化剂,发现了催化剂中心的Co2+先还原生成Co+,Co+再与CO2发生还原反应61。原位红外光谱发现B掺杂的催化剂在CO2还原中的中间介质是OOC―COO101。因此,原位表征技术为理解CO2还原过程提供了直接有效的方法。原位表征实验得出的数据与理论计算相结合可以进一步深入研究反应的中间体与催化剂表面原子结构之间的相互作用,活性位点的电子配体和催化行为。只有了解这些关键信息,才能设计出CO2还原的高性能电催化剂及其电催化体系。
(4) 新型催化剂体系
随着科技的不断发展,非金属催化剂越来越受到人们的关注,如,部分氧化4原子层厚度Co可以在低电位下进行CO2还原50。金属Sn超薄层嵌入到石墨烯层间结构中可以获得很高的甲酸电流效率,并能稳定运行长达50 h78。Gao等54发现Co3O4单元层结构中的O(II)空位存在有利于甲酸的生成。因此,对原子层厚度的非贵金属催化剂、金属超薄层嵌入石墨烯层间结构催化剂、金属催化剂的氧空位等新型体系的研究,有利于进一步提高CO2电催化还原效率。
综上所述,CO2电化学还原提供了一种将温室气体CO2转化为有价值的燃料的重要方法。利用可再生能源(如风能、水能等)提供的低品阶电力,驱动CO2电化学转化,从而实现有效的人工光合过程。这将开启一种新型可再生能源储存和CO2资源化转化利用的可持续发展模式。在CO2电化学还原的电催化剂材料,以及反应机理研究等方面,不断取得的重大突破,将使得CO2电化学还原技术在未来10年有望成为CO2转化利用的重要手段。
(1) Goeppert, A.; Czaun, M.; Jones, J. P.; Surya Prakash, G. K.; Olah, G. A.2014,, 7995. doi: 10.1039/c4cs00122b
(2) Windle, C. D.; Reisner, E.2015,, 435. doi: 10.2533/chimia.2015.435
(3) Pakhare, D.; Spivey, J.2014,, 7813. doi: 10.1039/c3cs60395d
(4) Kondratenko, E. V.; Mul, G.; Baltrusaitis, J.; Larrazábal, G. O.; Pérezramírez, J.2013,, 3112. doi: 10.1039/C3EE41272E
(5) Hanc-Scherer, F. A.; Montiel, M. A.; Montiel, V.; Herrero, E.; Sánchez-Sánchez, C. M.2015,, 23909. doi:10.1039/c5cp02361k
(6) http://djfj.renewable.org.cn (accessed March 31, 2017).
(7) Kortlever, R.; Shen, J.; Schouten, K. J. P.; Calle-Vallejo, F.; Koper, M. T. M.2015,, 4073. doi: 10.1021/acs.jpclett.5b01559
(8) Zhang, X.; Wu, Z. S.; Zhang, X.; Li, L. W.; Li, Y. Y.; Xu, H. M.; Li, X. X.; Yu, X. L.; Zhang, Z. S.; Liang, Y. Y.; Wang, H. L.2017,, 14675. doi: 10.1038/ncomms14675
(9) Appel, A. M.; Bercaw, J. E.; Bocarsly, A. B.; Dobbek, H.; Dubois, D. L.; Dupuis, M.; Ferry, J. G.; Fujita, E.; Hille, R.; Kenis, P. J. A.2013,, 6621. doi: 10.1021/cr300463y
(10) Lu Q, Rosen J,Jiao F., 2015,, 2. doi: 10.1002/cctc.201402669
(11) Lu, Q.; Rosen, J.; Zhou, Y.; Hutchings, G. S.; Kimmel, Y. C.; Chen, J. G.; Jiao, F.2014,, 3242. doi: 10.1038/ncomms4242
(12) Rosen, J.; Hutchings, G. S.; Lu, Q.; Rivera, S.; Zhou, Y.; Vlachos, D. G.; Jiao, F.2015,, 4293. doi: 10.1021/acscatal.5b00840
(13) Wang, Q. Q.; Chen, C. Z.; Zhong, J. H.; Zhang, B.; Cheng, Z. M.2016,, 293. doi: 10.1071/CH16138
(14) Ma, S.; Lan, Y.; Perez, G. M. J.; Moniri, S.;Kenis, P. J. A.2014,, 866. doi: 10.1002/cssc.201300934
(15) Hsieh, Y. C.; Senanayake, S. D.; Zhang, Y.; Xu, W.; Polyansky, D. E.2015,, 2584. doi: 10.1021/acscatal.5b01235
(16) Kim, D.; Resasco, J.; Yu, Y.; Asiri, A. M.; Yang, P.2014,, 4948. doi: 10.1038/ncomms5948
(17) Kortlever, R.; Peters, I.; Koper, S.; Koper, M. T. M.2015,, 3916. doi: 10.1021/acscatal.5b00602
(18) Rasul, S.; Anjum, D. H.; Jedidi, A.; Minenkov, Y.; Cavallo, L.; Takanabe, K.2014,, 2174. doi: 10.1002/ange.201410233
(19) Liu, Y.; Chen, S.; Quan, X.; Yu, H.2016,, 11631. doi: 10.1021/jacs.5b02975
(20) Varela, A. S.; Ranjbar, Sahraie N. Steinberg, J.; Ju, W.; Oh, H. S.;Strasser, P.2015,, 10908. doi: 10.1002/anie.201502099
(21) Asadi, M.; Kumar, B.; Behranginia, A.; Rosen, B. A.; Baskin, A.; Repnin, N.; Pisasale, D.; Phillips, P.; Zhu, W.; Haasch, R.2014,, 4470. doi: 10.1038/ncomms5470
(22) Nakata, K.; Ozaki, T.; Terashima, C.; Fujishima, A.; Einaga, Y.2014,, 890. doi: 10.1002/ange.201308657
(23) Hoang, T. H.; Ma, S.; Gold, J. I.; Kenis, P. J. A.; Gewirth, A. A.2017,, 3313. doi: 10.1021/acscatal.6b03613
(24) Rosen, J.; Hutchings, G. S.; Lu, Q.; Forest, R. V.; Moore, A.; Jiao, F.2015,, 4586. doi: 10.1021/acscatal.5b00922
(25) Kuhl, K. P.; Hatsukade, T.; Cave, E. R.; Abram, D. N.; Kibsgaard, J.; Jaramillo, T. F.2014,, 14107. doi: 10.1021/ja505791r
(26) Lum, Y.; Kwon, Y.; Lobaccaro, P.; Chen, L.; Clark, E. L.; Bell, A. T.; Ager, J. W.2015, 202. doi: 10.1021/acscatal.5b02399
(27) Zhang, S.; Kang, P.; Bakir, M.; Lapides, A. M.; Dares, C. J.; Meyer, T. J.2015,, 15809. doi: 10.1073/pnas.1522496112
(28) Li, F.; Zhao, S. F.; Chen, L.; Khan, A.; Macfarlane, D. R.; Zhang, J.2015,, 216. doi: 10.1039/C5EE02879E
(29) Li, C. W.; Kanan, M. W.2012,, 7231. doi: 10.1021/ja3010978
(30) Shen, J.; Kortlever, R.; Kas, R.; Birdja, Y. Y.; Diaz-Morales, O.; Kwon, Y.; Ledezma-Yanez, I.; Schouten, K. J. P.; Mul, G.; Koper, M. T. M.2015,: 8177. doi: 10.1038/ncomms9177
(31) Zhu, W.; Michalsky, R.; Metin, Ö.; Lv, H.; Guo, S.; Wright, C. J.; Sun, X.; Peterson, A. A.; Sun, S.2013,, 16833. doi: 10.1021/ja409445p
(32) Costentin, C.; Robert, M.; Saveant, J. M.2012,, 2423. doi: 10.1039/c2cs35360a
(33) Qu, Y.; Duan, X.2013,, 2568. doi: 10.1039/c2cs35355e
(34) Qiao, J.; Liu, Y.; Hong, F.; Zhang J.2013,, 631. doi: 10.1039/c3cs60323g
(35) Back, S.; Yeom, M. S.; Jung, Y.2015,, 5089. doi: 10.1021/acscatal.5b00462
(36) Baturina, O. A.; Lu, Q.; Padilla, M. A.; Xin, L.; Li, W.; Serov, A.; Artyushkova, K.; Atanassov, P.; Xu, F.; Epshteyn, A.; Brintlinger, T.; Schuette, M.;Collins, G. E.2014,, 3682. doi: 10.1021/cs500537y
(37) Zhu, W.; Zhang, Y. J.; Zhang, H.; Lv, H.; Li, Q.; Michalsky, R.; Peterson, A. A. Sun, S.2014,, 16132. doi: 10.1021/ja5095099
(38) Li, Q.; Sun, S.. 2016,, 178–197. doi: 10.1016/j.nanoen.2016.02.030
(39) Zhu, W.; Michalsky, R.; Lv, H.; Guo, S.; Wright, C. J.; Sun, X.; Peterson, A. A.; Sun, S.2013,, 16833. doi: 10.1021/ja409445p
(40) Gao, D.; Zhou, H.; Wang, J.; Miao, S.; Yang, F.; Wang, G.; Wang, J.; Bao, X.2015,, 4288. doi: 10.1021/jacs.5b00046
(41) Peterson, A. A.; Nørskov, J. K.. 2012,, 251. doi: 10.1021/jz201461p
(42) Hansen, H. A.; Varley, J. B.; Peterson, A. A.; Nørskov, J. K.2013,, 388. doi: 10.1021/jz3021155
(43) Luc, W.; Collins, C.; Wang, S.; Xin, H.; He, K.; Kang, Y.;Jiao, F.2017,, 1885. doi: 10.1021/jacs.6b10435
(44) Zhong, H.; Qiu, Y.; Zhang, T.; Li, X.; Zhang, H.;Chen, X.2016,, 13746. doi: 10.1039/C6TA06202D
(45) Lv, W.; Zhou, J.; Bei, J.; Zhang, R.; Wang, L.; Xu, Q.;Wang, W.2017,, 191. doi: 10.1016/j.apsusc.2016.10.017
(46) Chen, Y.; Li, C. W.; Kanan, M. W.2012,, 19969. doi: 10.1021/ja309317u
(47) Li, C. W.; Ciston, J.;Kanan, M. W.2014,, 504. doi: 10.1038/nature13249
(48) Chen, Y.;Kanan, M. W.2012,, 1986. doi: 10.1021/ja2108799
(49) Sun, Y.; Gao, S.; Lei, F.; Xiao, C.; Xie, Y.2015,, 3. doi: 10.1021/ar500164g
(50) Gao, S.; Lin, Y.; Jiao, X.; Sun, Y.; Luo, Q.; Zhang, W.; Li, D.; Yang, J.; Xie, Y.2016,, 68. doi: 10.1038/nature16455
(51) Zhang, S.; Kang, P.;Meyer, T. J.2014,, 1734. doi: 10.1021/ja4113885
(52) Gu, J. Wuttig, A.; Krizan, J. W.; Hu. Y.; Detweiler, Z. M.; Cava, R. J.; Bocarsly, A. B.2013,, 12415. doi: 10.1021/jp402007z
(53) Watkins, J. D.; Bocarsly, A. B.. 2014,, 284. doi: 10.1002/cssc.201300659
(54) Gao, S.; Sun, Z.; Liu, W.; Jiao, X.; Zu, X.; Hu, Q.; Sun, Y.; Yao, T.; Zhang, W.; Wei, S.; Xie, Y.2017,, 14503. doi: 10.1038/ncomms14503
(55) Mistry, H.; Varela, A. S.; Bonifacio, C. S.; Zegkinoglou, I.; Sinev, I.; Choi, Y. W.; Kisslinger, K.; Stach, E. A.; Yang, J. C.; Strasser, P.; Cuenya, B. R.2016,, 12123. doi: 10.1038/ncomms12123
(56) Chen, L.; Guo, Z.; Wei, X. G.; Gallenkamp, C.; Bonin, J.; Anxolabéhère-Mallart, E.; Lau, K. C.; Lau, T. C.; Robert, M.2015,, 10918. doi: 10.1021/jacs.5b06535
(57) Costentin, C.; Savéant, J. M.2012,, 90. doi: 10.1126/science.1224581
(58) Yao, S. A.; Ruther, R. E.; Zhang, L.; Franking, R. A.; Hamers, R. J.; Berry, J. F.2017,, 15632. doi: 10.1021/ja304783j
(59) Tornow, C. E.; Thorson, M. R.; Ma, S.; Gewirth, A. A.; Kenis, P. J.2012,, 19520. doi: 10.1021/ja308217w
(60) Lin, S.; Diercks, C. S.; Zhang, Y. B.; Kornienko, N.; Nichols, E. M.; Zhao, Y.; Paris, A. R.; Kim, D.; Yang, P.; Yaghi, O. M.2015,, 1208.
(61) Hod, I.; Farha, O. K.; Hupp J. T.2015,, 1192. doi: 10.1038/nmat4494
(62) Kornienko, N.; Zhao, Y.; Kley, C. S.; Zhu, C.; Kim, D.; Lin, S.; Chang, C. J.; Yaghi, O. M.; Yang, P.2015,, 14129. doi: 10.1021/jacs.5b08212
(63) Hod, I.; Sampson, M. D.; Deria, P.; Kubiak, C. P.; Farha, O. K.; Hupp, J. T.2015,, 6302. doi: 10.1021/acscatal.5b01767
(64) Hinogami, R.; Yotsuhashi, S.; Deguchi, M.; Zenitani, Y.; Hashiba, H.; Yamada, Y.2012,, 17. doi: 10.1149/2.001204eel
(65) Senthil, K. R.; Senthil, Kumar S.; Anbu, Kulandainathan M.2012,, 70. doi: 10.1016/j.elecom.2012.09.018
(66) Kang, X.; Zhu, Q.; Sun, X.; Hu, J.; Zhang, J.; Liu, Z.;Han, B.2016,, 266. doi: 10.1039/C5SC03291A
(67) Roberts, F. S.; Kuhl, K. P.; Nilsson, A.2015,, 5179. doi: 10.1002/anie.201412214
(68) Kas, R.; Kortlever, R.; Milbrat, A.; Koper, M. T. M.; Mul, G.; Baltrusaitis, J.2014,, 12194. doi: 10.1039/c4cp01520g
(69) Kuhl, K. P.; Cave, E. R.; Abram, D. N.; Jaramillo, T. F.2012,, 7050. doi: 10.1039/C2EE21234J
(70) Zhang, Z.; Qi, Z. M.; Zhang, R. J.2012,, 1163. [张 喆, 祁志美, 张蓉君. 物理化学学报, 2012,, 1163.] doi: 10.3866/PKU.WHXB201202241
(71) Zuo, K.; Liang, S.; Liang, P.; Zhou, X.; Sun, D.; Zhang, X.; Huang, X.2015,, 426. doi: 10.1016/j.biortech.2015.02.108
(72) Katuri, K. P.; Werner, C. M.; Jimenez-Sandoval, R. J.; Chen, W.; Jeon, S.; Logan, B. E.; Lai, Z.; Amy, G. L.; Saikaly, P. E.2014,, 12833. doi: 10.1021/es504392n
(73) Kas, R.; Hummadi, K. K.; Kortlever, R.; de, Wit P.; Milbrat, A.; Luiten-Olieman, M. W. J.; Benes, N. E.; Koper, M. T. M.; Mul, G.2016,, 10748. doi: 10.1038/ncomms10748
(74) Chen, S.; Duan, J.; Ran, J.; Jaroniec, M.; Qiao, S. Z.2013,, 3693. doi: 10.1039/C3EE42383B
(75) Gao, S.; Jiao, X.; Sun, Z.; Zhang, W.; Sun, Y.; Wang, C.; Hu, Q.; Zu, X.; Yang, F.;Yang, S.2016,, 708. doi: 10.1002/anie.201509800
(76) Liang, L.; Lei, F.; Gao, S.; Sun, Y.; Jiao, X.; Wu, J.; Qamar, S.; Xie, Y.2015,, 13971. doi: 10.1002/anie.201506966
(77) Deng, J.; Ren, P.; Deng, D.; Bao, X.2015,, 2100. doi: 10.1002/anie.201409524
(78) Lei, F.; Liu, W.; Sun, Y.; Xu, J.; Liu, K.; Liang, L.; Yao, T.; Pan, B.; Wei, S.; Xie, Y.2016,, 12697. doi: 10.1038/ncomms12697
(79) Wu, J.; Yadav, R. M.; Liu, M.; Sharma, P. P.; Tiwary, C. S.; Ma, L.; Zou, X.; Zhou, X-D; Yakobson, B. I.; Lou, J.; Ajayan, P. M.2015,, 5364. doi: 10.1021/acsnano.5b01079
(80) Wu, J.; Liu, M.; Sharma, P. P.; Yadav, R. M.; Ma, L.; Yang, Y.; Zou, X.; Zhou, X-D; Vajtai, R.; Yakobson, B. I.; Lou, J.; Ajayan, P. M.2016,, 466. doi: 10.1021/acs.nanolett.5b04123
(81) Kumar, B.; Asadi, M.; Pisasale, D.; Sinha-Ray, S.; Rosen, B. A.; Haasch, R.; Abiade, J.; Yarin, A. L.; Salehi-Khojin, A.2013,, 2819. doi: 10.1038/ncomms3819
(82) Zhang, S.; Kang, P.; Ubnoske, S.; Brennaman, M. K.; Song, N.; House, R. L.; Glass, J. T.; Meyer, T. J.2014,, 7845. doi: 10.1021/ja5031529
(83) Sreekanth, N.; Nazrulla, M. A.; Vineesh, T. V.; Sailaja, K.; Phani, K. L.2015,, 16061. doi: 10.1039/c5cc06051f
(84) Wu, J.; Ma. S.; Sun, J.; Gold, J. I.; Tiwary, C.; Kim, B.; Zhu, L.; Chopra, N.; Odeh, I. N.; Vajtai, R.; Yu, A. Z.; Luo, R.; Lou, J.; Ding, G.; Kenis, P. J. A.; Ajayan, P. M.2016,, 13869. doi: 10.1038/ncomms13869
(85) Prakash, G. K. S.; Viva, F. A.; Olah, G. A.2013,, 68. doi: 10.1016/j.jpowsour.2012.09.036
(86) Seok, Ki K.; Yin-Jia, Z.; Helen, B.; Ronald, M.; Andrew, P.2016,, 2003. doi: 10.1021/acscatal.5b02424.s001
(87) Hatsukade, T.; Kuhl, K. P.; Cave, E. R.; Abram, D. N.; Jaramillo, T. F.2014,, 13814. doi: 10.1039/C4CP00692E
(88) Alves, D. C. B.; Silva, R.; Voiry, D.; Asefa, T.; Chhowalla, M.2015,, 2. doi: 10.1007/s40243-015-0042-0
(89) Baruch, M. F.; Pander, J. E.; White, J. L.; Bocarsly, A. B.2015,, 3148. doi: 10.1021/acscatal.5b00402
(90) Fang, Y.; Flake, J. C.2017,, 3399. doi: 10.1021/jacs.6b11023
(91) Morais, J. P.; Rosa, M. F.; De, S. F. M. S.; Nascimento, L. D.; do Nascimento, D. M.; Cassales, A. R.2013,, 229. doi: 10.1016/j.carbpol.2012.08.010
(92) Shi, L.; Liu, Q.; Guo, X.; Wu, W.; Liu, Z.2013,, 125. doi: 10.1016/j.fuproc.2012.06.023
(93) Puthiyapura, V. K.; Dan, J. L. B.; Russell, A. E.; Lin, W. F.; Hardacre, C.2016,, 12859. doi:10.1021/acsami.6b02863
(94) Calle-Vallejo, F.; Koper, M. T. M.2012,, 3. doi: 10.1016/j.electacta.2012.04.062
(95) Wu, Z.; Lv, Y.; Xia, Y.; Webley, P. A.; Zhao, D.2012,, 2236. doi: 10.1021/ja209753w
(96) Kong, B.; Selomulya, C.; Zheng, G.; Zhao, D.2015,, 7997. doi: 10.1039/c5cs00397k
(97) Tang, J.; Liu, J.; Torad, N. L.; Kimura, T.; Yamauchi, Y.2014,, 305. doi: 10.1016/j.nantod.2014.05.003
(98) Malgras, V.; Ataee-Esfahani, H.; Wang, H.; Jiang, B.; Li, C.; Wu, K. C. W.; Kim, J. H.; Yamauchi, Y.2016,, 993. doi: 10.1002/adma.201502593
(99) Hall, A. S.; Yoon, Y.; Wuttig, A.; Surendranath, Y.2015,, 14834. doi: 10.1021/jacs.5b08259
(100) Jones, J. P.; Prakash, G. K. S.; Olah, G. A.2014,, 1451. doi: 10.1002/ijch.201400081
(101) Liu, Y.; Chen, S.; Quan, X.; Yu, H.2015,, 11631. doi: 10.1021/jacs.5b0297
Recent Progress on Electrochemical Reduction of Carbon Dioxide
BAI Xiao-Fang1,2CHEN Wei2,*WANG Bai-Yin1,2FENG Guang-Hui1,2WEI Wei2JIAO Zheng1SUN Yu-Han2,*
(1;2)
Conversion of carbon dioxide (CO2) to value-added chemicals and fuels driven by low-grade renewable electricity is of significant interest since it serves the dual purpose of reducing atmospheric content of CO2by utilizing it as a feedstock and storing it in the form of high-energy-density fuels. In this regard, there are an increasing number of interesting developments taking place in the popular research focus area of electrochemical reduction of CO2. This review first introduces the general principles of CO2electroreduction. Next, the latest progress relating to electrocatalytic materials and experimental and theoretical studies of the reaction mechanism has been discussed. Finally, the challenges and prospects for further development of CO2electroreduction have been presented.
Carbondioxide; Renewableenergy; Electrochemicalreduction; Electrocatalyst; Reactionmechanism
April 10, 2017;
May 29, 2017;
June 13, 2017.
Corresponding authors.CHEN Wei, Email: chenw@sari.ac.cn. SUN Yu-Han, Email: sunyh@sari.ac.cn; Tel: +86-21-2035-0997.
10.3866/PKU.WHXB201706131
O646
The project was supported by the Ministry of Science and Technology, China (2016YFA0202800) and the Hundred Talents Program of Chinese Academy of Sciences, China.
科技部国家重点研发计划(2016YFA0202800)和中国科学院百人计划项目资助
