基于形态学与核糖体DNA及其cDNA高通量测序的海洋沉积物中纤毛虫多样性比较*
2018-01-12黄平平徐奎栋
黄平平 赵 峰 徐奎栋①
(1. 中国科学院海洋研究所 海洋生物分类与系统演化实验室 青岛 266071; 2. 中国科学院大学 北京 100049)
底栖纤毛虫作为底栖微食物网中的重要成员能够促进初级生产力向更高营养级转换, 从而促进底栖生态系统物质循环和能量流动(徐奎栋, 2011)。过去对海洋底栖纤毛虫多样性的认识主要基于活体观察或固定染色的形态学研究方法。例如 Hausmann等(2002)通过半定量培养的方法, 对地中海东部海域底栖原生动物进行研究, 共发现35种纤毛虫。在黄海,Meng等(2012)通过密度梯度离心-定量蛋白银染色法(Ludox-QPS)对夏季的48个站位进行了分析, 共鉴定198种底栖纤毛虫; 代仁海(2012)以相同方法在春季黄海的11个站位获得96种底栖纤毛虫。然而, 形态学研究手段自身存在的问题和不足限制了对底栖纤毛虫多样性更为深入的认识, 如耗时费力, 需要良好的分类学基础, 大多数类群个体数量少、个体微小且难以培养而易被忽视, 未有针对包囊的分类学依据,固定液的效能会导致不同程度的低估等(Karayanni et al, 2004; 陈旭淼等, 2014)。
与传统形态学研究方法相比, 基于基因测序的分子生物学研究手段突破了对纤毛虫个体数量及培养的限制, 理论上只要有遗传物质存在即可被检测到。目前高通量测序技术的发展极大地扩展了对海洋底栖纤毛虫多样性的认识。Gong等(2015)通过 18S rRNA基因焦磷酸测序对黄海底栖真核微生物的研究中发现, 就序列数和可操作分类单元(operational taxonomic units, OTUs)数所占比例而言, 甲藻和纤毛虫均为优势类群。Pawlowski等(2011)以相同方法, 在不足4g的沉积物中检获630余个纤毛虫OTUs。然而,通过形态学手段却无法获得如此众多的物种。PCR和测序等过程均可能导致对多样性的高估, 但目前对其高估程度还难以准确定量(Kunin et al, 2010)。此外,不同的数据处理软件、参数的选择等均可导致OTUs数的改变, 进而影响分子多样性的评价(Santoferrara et al, 2014)。由此, 通过分子手段检获的这种高多样性是真实存在, 还是由于分子技术和数据处理等问题造成的假象, 依然不得而知。
鉴于形态学研究手段以及分子生物学技术在揭示纤毛虫多样性方面均存在一定的问题与不足, 结合多个方法进行比较研究, 探讨不同技术手段获得结果的异同, 显得尤为重要。研究者们曾以易于鉴定的浮游丁丁虫类(Tintinnid)纤毛虫为研究对象, 比较分析形态观察和 DNA测序两种方法获得的多样性,发现分子手段检获的多样性较形态学方法高一到两个数量级(Bachy et al, 2013; Santoferrara et al, 2014)。然而上述研究仅涉及单一的浮游类群。
迄今, 尚没有工作涉及沉积物中纤毛虫的方法学比较。沉积物环境不仅包括底栖纤毛虫活动虫体的遗传信息, 还包含浮游为主的寡毛亚纲和舞毛亚纲纤毛虫(Grattepanche et al, 2014)裂解释放的DNA以及它们的包囊等遗传物质(Doherty et al, 2010; Torti et al, 2015)。Reid 等(1978)和 Rubino 等(2000)研究了沉积物中浮游类群的包囊, 发现了浮游丁丁虫类纤毛虫包囊的存在。Kim等(2008)对日本女川湾及韩国马山湾沉积物中浮游类纤毛虫(具头急游虫)包囊的形态及其原位沉降的研究中发现, 在一定条件下, 浮游类纤毛虫的包囊会以较大速率沉降。因此, 基于 DNA测序检获的沉积物中的纤毛虫多样性, 可能既包括活动虫体, 同时也包括胞外 DNA和以包囊形式存在的纤毛虫。与DNA相比, RNA降解速率较快(Novitsky,1986), 因此通过 cDNA测序, 理论上可获取纤毛虫活动虫体的多样性。
本文以海洋沉积物中的纤毛虫为研究对象, 通过 Ludox-QPS制片, 显微观察获取活动虫体的多样性构成(Xu et al, 2010), 同时提取沉积物中总DNA/RNA, 测序获得纤毛虫分子多样性构成信息,对上述三种方法获得的结果进行比较分析, 探讨基于形态学与核糖体18S DNA及其cDNA高通量测序手段检获的海洋沉积物中纤毛虫多样性的异同, 为后续分子多样性研究提供理论依据。
1 材料与方法
1.1 调查站位与样品采集
本研究于2015年8月17日搭乘“东方红2号”科学考察船在南黄海(36°0′3.6″N, 121°20′27.6″E)进行样品采集, 水深 34.91m, 底层水温度 12.87°C, 底层水盐度31.52。
利用0.1m2改进型Gray-Ohara箱式采泥器采集2箱未受扰动的沉积物样品: (1) 使用注射器改造的采样管(内径2.3cm)采集0—2cm芯样分别进行瓶装, 加入等体积预冷 4%戊二醛进行固定, 置于 4°C冷藏保存; (2) 同时刮取0—2cm表层沉积物约20g放入封口袋中, 快速置于-20°C冰箱中冷冻保存。
1.2 形态学观察
采用Ludox-QPS法分析纤毛虫的物种多样性(Xu et al, 2010), 共两个生物学重复(两箱泥), 各一个技术重复。主要步骤为: 采集的2个固定的沉积物样品首先进行降盐浓缩, 每一重复取9mL(经验值为3mL,但为确保多样性数据准确, 采用 9mL使每张片子上的纤毛虫数均达到200以上)样品加入Ludox HS 40密度梯度离心, 取生物层过滤到孔径 5µm 的硝酸纤维素膜上, 定量蛋白银染色制片(QPS), 最终两张片子(两箱泥)在 200-1000×(Leica DM 4500B)放大倍数下全片镜检观察。依据 Carey(1992), Lynn等(2002),Lynn(2008), 宋微波等(2009)及相关的文献资料, 大多数纤毛虫鉴定到属, 部分鉴定到种。纤毛虫的绝对丰度以每立方厘米的纤毛虫虫数表示(cell/cm3), 相对丰度以每类群绝对丰度占总丰度的百分比表示。
1.3 DNA和RNA提取及PCR扩增
DNA提取和RNA反转录均采用两个生物学重复(两箱泥)、各3个技术重复。其中沉积物DNA提取采用PowerSoil DNA Isolation kit (Mo-Bio, USA), 6个样品(两箱泥, 每箱3个重复), 各取0.3g用于DNA提取; RNA提取采用 RNA PowerSoil Total RNA Isolation kit (Mo-Bio, USA), 2个样品(两箱泥, 每箱1个), 各取2g用于RNA提取。每箱泥提取的总RNA取 3个分样, 通过 PrimerScript Ⅱ1st strand cDNA Synthesis Kit反转录成cDNA, 提取的DNA以及反转录合成的cDNA通过巢式PCR对纤毛虫18S rRNA基因V4区进行特异性扩增(Stock et al, 2013), 扩增引物参照文献(Lara et al, 2007; Stoeck et al, 2010): 纤毛虫特异性引物Cil F、Cil R1、Cil R2、Cil R3; 真核V4高变区通用引物EukF、EukR(表1)。

表1 纤毛虫巢式PCR特异性引物及碱基序列表Tab.1 Oligonucleotide sequences and ciliate-specific primers used for the ciliate nested-PCR
第一轮PCR: 采用纤毛虫特异性引物, 扩增长度600余bp, 反应体系如下: 正反向引物各0.5µL, 模板1.5µL, 2×TransTaq High Fidelity PCR Supermix 12.5µL, 最后加双蒸水补齐 25µL。反应流程: 95°C 预变性 5min; 然后 94°C变性 45s, 58°C退火 1min, 72°C延伸1min, 共35个循环; 最后72°C延伸10min终止于 12°C(Laraet al, 2007)。
第二轮 PCR: 以第一轮 PCR产物为模板, 采用真核特异性引物对 V4高变区进行特异性扩增, 扩增长度约 400bp, 反应体系如下: 正反向引物各 1µL,模板 1.5µL, 2×TransTaq High Fidelity PCR Supermix 23.5µL, 最后加双蒸水补齐 50µL。反应流程: 95°C 预变性 5min; 然后 94°C 变性 30s, 57°C 退火 45s, 72°C延伸1min, 共10个循环; 94°C变性30s, 49°C退火45s,72°C延伸1min, 共25个循环; 最后72°C延伸2min终止于 12°C(Stoecket al, 2010)。
1.4 测序及序列数据分析
琼脂糖凝胶电泳检测 PCR产物质量, 若符合要求, 则将来自同一箱泥的3个重复的核糖体18S DNA及其 cDNA的 PCR产物分别进行合并, 最终核糖体18S DNA及其cDNA的PCR产物各两组(两箱泥), 进行Illumina Miseq测序(下简称DNA测序和cDNA测序)。
样品送测北京诺禾致源生物信息科技有限公司,使用New England Biolabs公司的NEB Next UltraTMDNA Library Prep Kit for Illumina建库试剂盒进行文库的构建, 构建好的文库经过Qubit定量和文库检测,合格后, 使用Illumina MiSeq进行上机测序。
根据 Barcode序列和PCR扩增引物序列从下机数据中拆分出样品数据, 截去 Barcode和引物序列,使用FLASH(V1.2.7)对样品reads(DNA测序两重复共209055 reads, cDNA测序两重复共90706 reads)进行拼接, 得到的拼接序列为原始Tags数据即Raw Tags(Magočet al, 2011)。采用 QIIME(V1.7.0)对序列进行过滤处理: a) Tags截取: 将Raw Tags从连续低质量值(默认质量阈值为≤19)碱基数达到设定长度(默认长度值为3)的第一个低质量碱基位点截断; b) Tags长度过滤: Tags经过截取后得到的Tags数据集, 进一步过滤掉其中连续高质量碱基长度小于Tags长度75%的Tags(Caporasoet al, 2010)。经以上处理得到的Tags序列通过 UCHIME与数据库(Gold Database)进行比对, 检测及去除嵌合体序列, 得到最终的有效数据即Effective Tags (Edgaret al, 2011; Haaset al, 2011)。
采用UPARSE(Edgar, 2013)对以上所得Effective Tags进一步处理。流程如下: 去重复, 统计每条序列丰度; 去除单一重复的序列; 以 97%水平进行 OTU聚类, 即97%的序列相似度视作同种。对获得的OTU代表序列与美国国家生物技术中心(National Center for Biotechnology Information, NCBI)数据库采用基本局部比对搜索工具(Basic Local Alignment Search Tool,BLAST)进行比对, 获得相应序列的分类信息。其中纤毛虫的相对丰度以每个类群的序列数占总序列数的比例表示。
1.5 数据统计分析
使用 Venny2.1绘制饼图, 使用 Excel绘制条形图。使用PRIMER软件包中的CLUSTER分析三种方法在揭示纤毛虫多样性及相对丰度方面的一致性;相似性百分比(Similarity Percentages, SIMPER)分析找出对聚类分组起主要作用的科, 分析之前原始数据进行log(x+1)转化。
2 结果与分析
2.1 形态观察、DNA及其cDNA测序所获纤毛虫多样性构成
三种分析方法中, 形态学方法检获的纤毛虫多样性最低, 包含8纲、20目、30科、36属、97种, 其中核残迹纲的部分类群、膜口亚纲的全部、旋唇纲和篮口纲的少数个体仅能鉴定到目级水平。以97%的相似性视为一个OTU, 则DNA测序检获的多样性次之,包含10纲、28目、55科、76属、174个OTUs。cDNA测序检获的多样性最高包含10纲、31目、68科、99属、284个OTUs。三种方法同时检获的有8纲、16目、20科、17属, 分别占形态学方法检获总数的100%、80%、67%、47%。而同时被两种分子手段检获的个数依次为10纲、28目、51科、72属, 在纲和目级水平上完全一致, 而在科属水平上分别占 DNA测序方法检获总数的93%和95%(图1)。
在属级阶元上, 三种方法同时检获的有 17属,包含形态鉴定获得的 30种纤毛虫, 占鉴定的纤毛虫总物种数的31%(图2)。该17属中, DNA及其cDNA测序分析获得的分别有32个和50个OTUs, 均占各自检获的总OTUs数的18%(图2)。此外, 另有17个属仅通过形态学鉴定获得, 而 84个属仅通过分子手段检获, 显示DNA及其cDNA测序方法获得了更多的属(图 1)。
对仅通过形态鉴定获得的17属的相关序列分析发现, 其中的 7个属包括丰富度较高的裸口虫属(Holophrya)和中圆虫属(Metacystis)在NCBI中均无相关序列。对仅通过分子检获的 84个属重新 BLAST比对发现, 在序列之间相似性十分近似甚至在同等相似下, 同一 OTU可以与多个属包括 5个仅通过形态学检测的属匹配。另外的5个形态检测到的属未能通过分子手段获得, 可能是个体数量少所致。其中,个体数量最少的盖雷虫属(Geleia)、双眉虫属(Diophrys)和腹毛虫属(Hypotrichidium)数量仅占纤毛虫总数的 0.24%, 而圆纤虫属(Strongylidium)个体数量仅占总数的 0.48%, 拟斜管虫属(Chilodontopsis)数量高一些, 占2.88%。
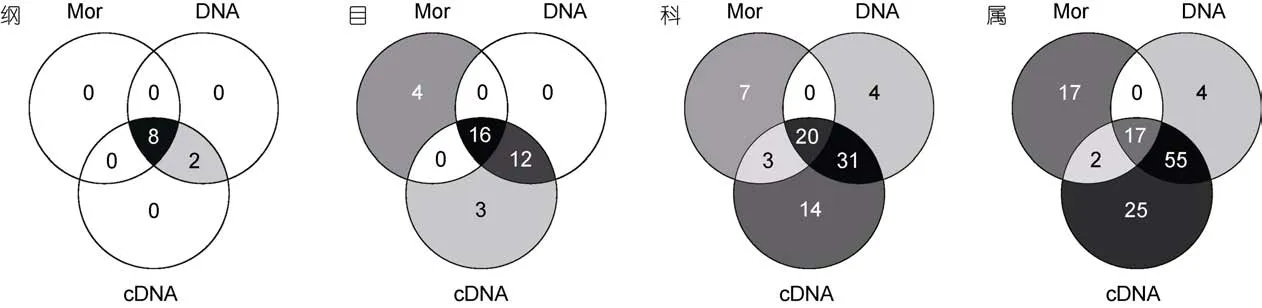
图1 形态学(Mor)、DNA及其cDNA测序三种方法在纲、目、科、属水平上检获的共有/特有的分类阶元数Fig.1 The number of shared and unique taxa at the levels of class/order/family/genus detected by morphological (Mor) and DNA and cDNA high-throughput methods

图2 形态学(Mor)、DNA及其cDNA测序三种方法检获的共有属的物种数/OTU数Fig.2 The number of morphospecies/OTUs at the level of genus simultaneously detected by morphological (Mor) and DNA and cDNA high-throughput methods
相对纤毛虫属级阶元, 科级阶元在NCBI数据库中更为完备。对三种方法检获的科级阶元的物种数/OTU数的聚类分析结果显示, 形态学的两个重复聚为一支(GroupⅠ), cDNA测序的两个重复聚为一支(Group Ⅲ),然后与 DNA 测序的两个重复(Group Ⅱ)聚在一起(图3a)。SIMPER分析表明, GroupⅠ与GroupⅡ的平均非相似性为 67%, GroupⅠ与 Group Ⅲ的平均非相似性为68%, Group Ⅱ与 Group Ⅲ的平均非相似性为 33%, 显示两种分子手段的结果更为近似。对形态学方法及DNA测序方法非相似性贡献率最高的科为中圆虫科,该科仅通过形态学方法检获, 主要贡献的属为中圆虫属(NCBI中无序列)丰富度位居第二; 仅通过DNA测序方法检获的浮游类群的累积贡献率高达 19%, 涉及 32个OTUs(表2)。对形态学方法及cDNA测序方法非相似性贡献率最高的科为刀口虫科, 其次为中圆虫科; 仅通过 cDNA测序方法检获的浮游类群的累积贡献率为12%, 寄生生活的无口类和后口类及厌氧生的斜毛类累积贡献率为12%, 共涉及65个OTUs(表3)。
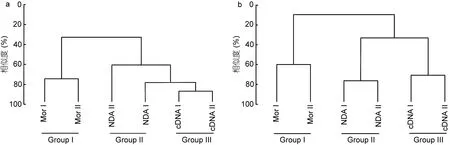
图3 形态学(Mor)、DNA及其cDNA测序三种方法检获的基于科级水平的物种数/OTUs (a)及其相对丰度(b)的聚类分析图Fig.3 Cluster analysis of the number of morphospecies/OTUs (a) at the level of family as well as their relative abundance (b) detected by morphological (Mor) and DNA and cDNA high-throughput methods
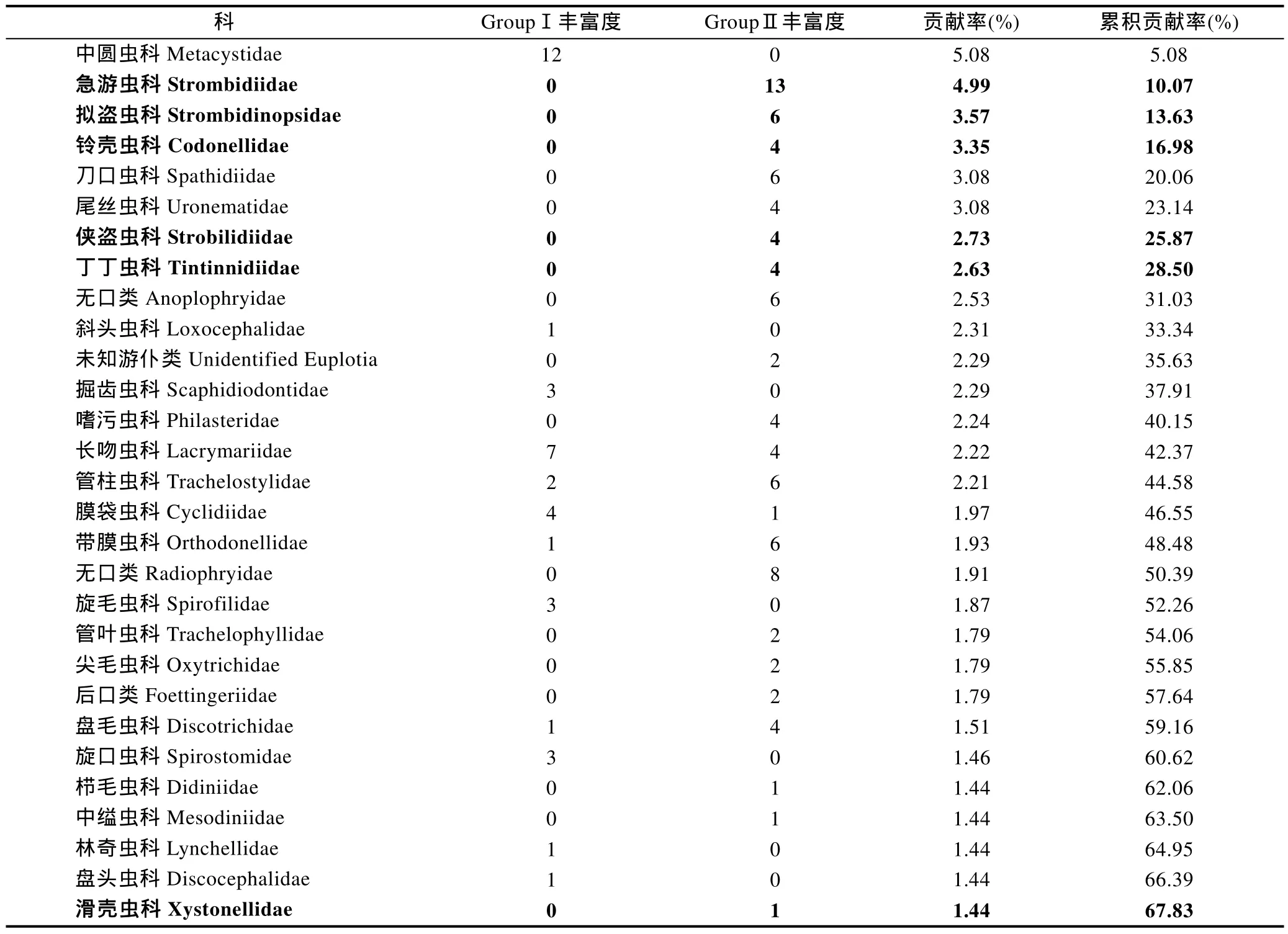
表2 基于科级水平丰富度聚类获得的形态组(GroupⅠ)和DNA测序组(GroupⅡ)的非相似性贡献(粗体示浮游类群)Tab.2 Dissimilarity contribution of the ciliate species richness at the level of family between Groups Ⅰ and Ⅱ (pelagic ciliates are indicated in boldface)
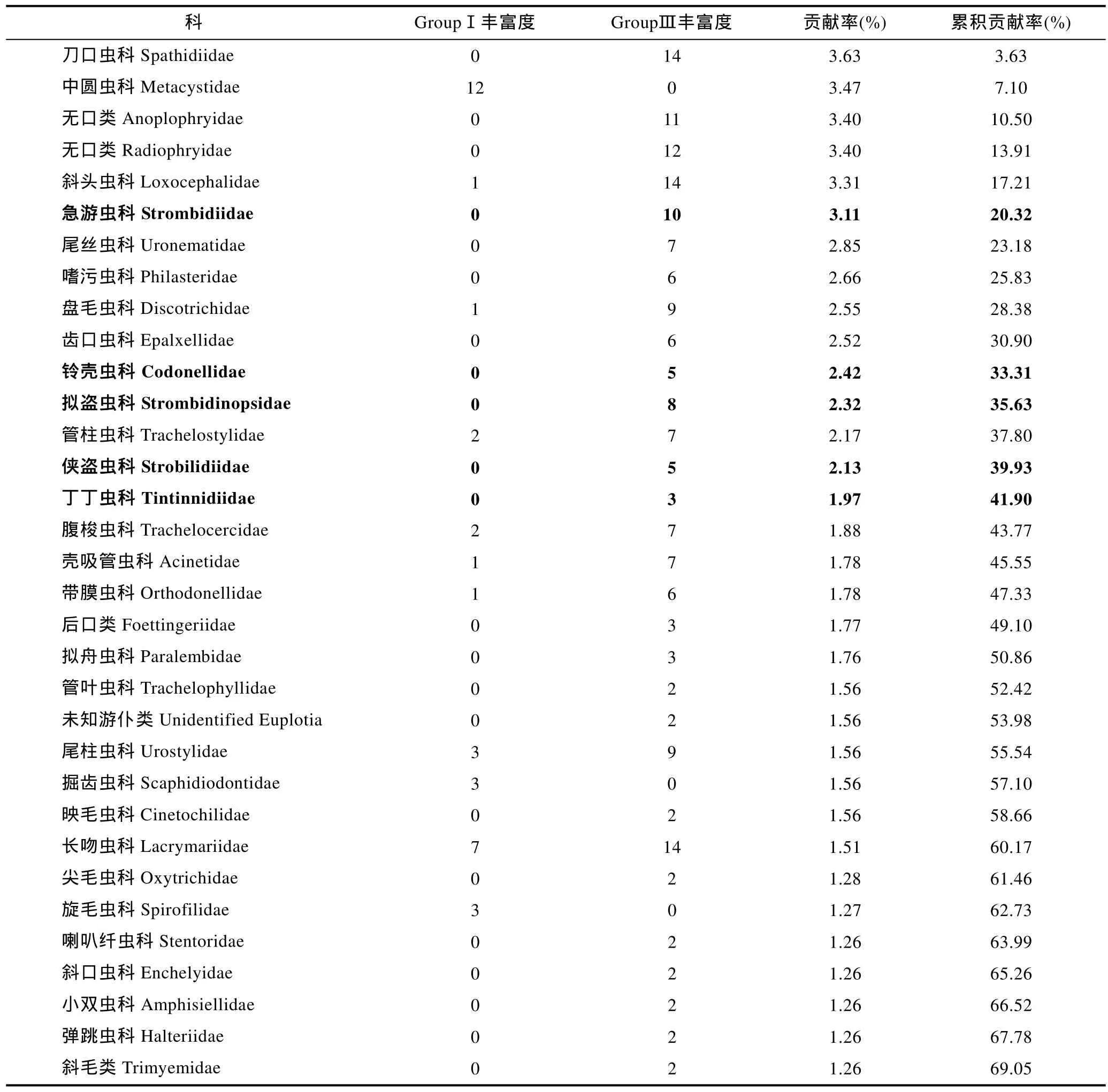
表3 基于科级水平丰富度聚类获得的形态组(GroupⅠ)和cDNA测序组(Group Ⅲ)的非相似性贡献(粗体示浮游类群)Tab.3 Dissimilarity contribution of the ciliate species richness at the level of family between Groups I and III (pelagic ciliates are indicated in boldface)
2.2 形态观察、DNA及其cDNA测序所获纤毛虫丰度构成
对科级阶元的优势类群分析表明, 形态学方法检获丰度最高的科为裸口虫科, 其中裸口虫属(Holophrya)贡献总丰度的38%; 中圆虫科居次, 其中圆虫属(Metacystis)贡献总丰度的13%。两属分别隶属前口纲的前管目和前口目, 均为黄海底栖生优势类群。形态学方法检获的均为底栖生类群。
DNA测序检获的相对丰度最高的科为钩刺亚纲(Haptoria)的栉毛虫科, 其中栉毛虫属(Cyclotrichium)为典型的浮游类群, 贡献了相对丰度的45%; 隶属于寡毛亚纲(Oligotrichia)的急游虫科、舞毛亚纲(Choreotrichia)的铃壳虫科和拟盗虫科亦属浮游纤毛虫, 共贡献了相对丰度的42%。值得注意的是, DNA检测的浮游纤毛虫占了纤毛虫总相对丰度的90%。
cDNA测序方法检获的相对丰度最高的科与形态学方法相同为裸口虫科, 其中的隐核虫属(Cryptocaryon)为寄生类纤毛虫, 贡献了相对丰度的22%。这可能是由于NCBI并不存在裸口虫属的序列,通过序列比对只能比对到较为相似的隐核虫属。隶属于钩刺亚纲的旋前管虫科相对丰度居次, 其中的伸颈虫属(Trachelotractus)贡献了相对丰度的 19%。cDNA方法检获的浮游类纤毛虫相对丰度为 7%。但就优势类群而言, cDNA方法与形态学方法更为相似,均为底栖生纤毛虫。
对形态观察、DNA及其cDNA测序三种方法所获科级纤毛虫相对丰度的聚类分析结果显示, 形态学检测的两个重复聚为一支(GroupⅠ), 然后与cDNA 测序的两个重复(GroupⅢ)聚在一起, DNA 测序的两个重复聚为另一支(GroupⅡ) (图3b)。SIMPER分析表明, GroupⅠ与GroupⅡ的平均非相似性为96%,GroupⅡ与GroupⅢ的平均非相似性为85%, GroupⅠ与GroupⅢ的平均非相似性为67%, 显示形态学方法与cDNA测序方法的结果更为近似。对形态学方法及DNA方法非相似性贡献的主要为浮游类纤毛虫, 仅通过 DNA方法检获, 累积贡献率为 48%, 涉及相对丰度的 90%; 裸口虫科和中圆虫科的累积贡献率为25%, 形态学方法检获了相对丰度的 51%, 而 DNA方法仅检获了2%(表4)。对DNA及其cDNA方法非相似性贡献的主要类群同样是浮游纤毛虫, 其累积贡献率为45%(表5)。对形态学方法及cDNA方法非相似性贡献的类群除急游虫科外, 均为底栖类群。造成差异的主要原因在于部分类群如中圆虫科(中圆虫属NCBI无序列)仅通过形态学方法检获, 贡献率为10%;其他类群如旋前管虫科、管叶虫科和刀口虫科仅通过cDNA方法检获, 累积贡献率为20%(表6)。
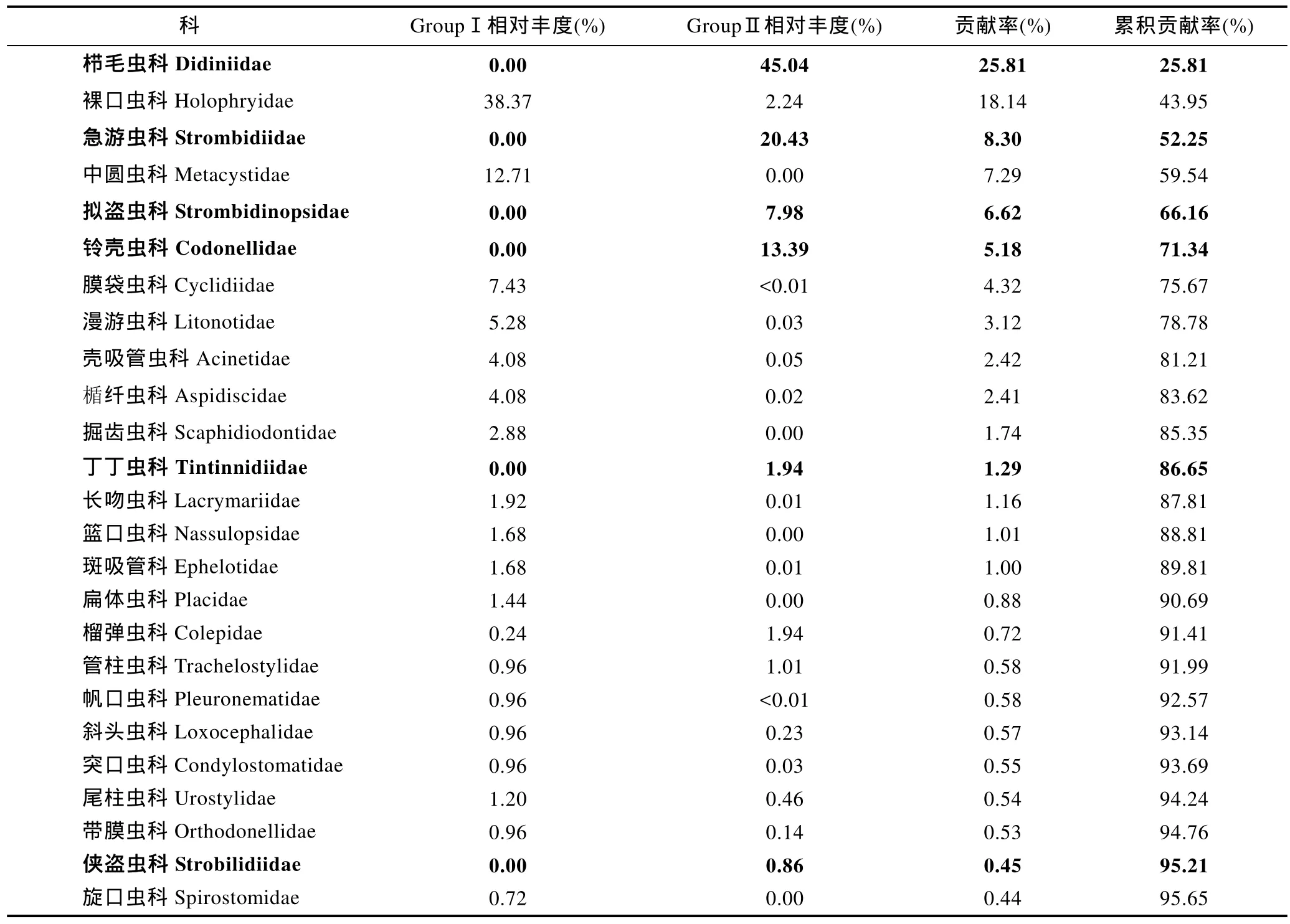
表4 基于科级水平相对丰度聚类获得的形态组(GroupⅠ)和DNA测序组(GroupⅡ)的非相似性贡献(粗体示浮游类群)Tab.4 Dissimilarity contribution of the ciliates species relate abundance at the level of family between Groups Ⅰ and Ⅱ (pelagic ciliates are indicated in boldface)

表5 基于科级水平相对丰度聚类获得的DNA(GroupⅡ)及其cDNA测序组(Group Ⅲ)的非相似性贡献(粗体示浮游类群)Tab.5 Dissimilarity contribution of the ciliates species relate abundance at the level of family between GroupsⅡ and Ⅲ (pelagic ciliates are indicated in boldface)
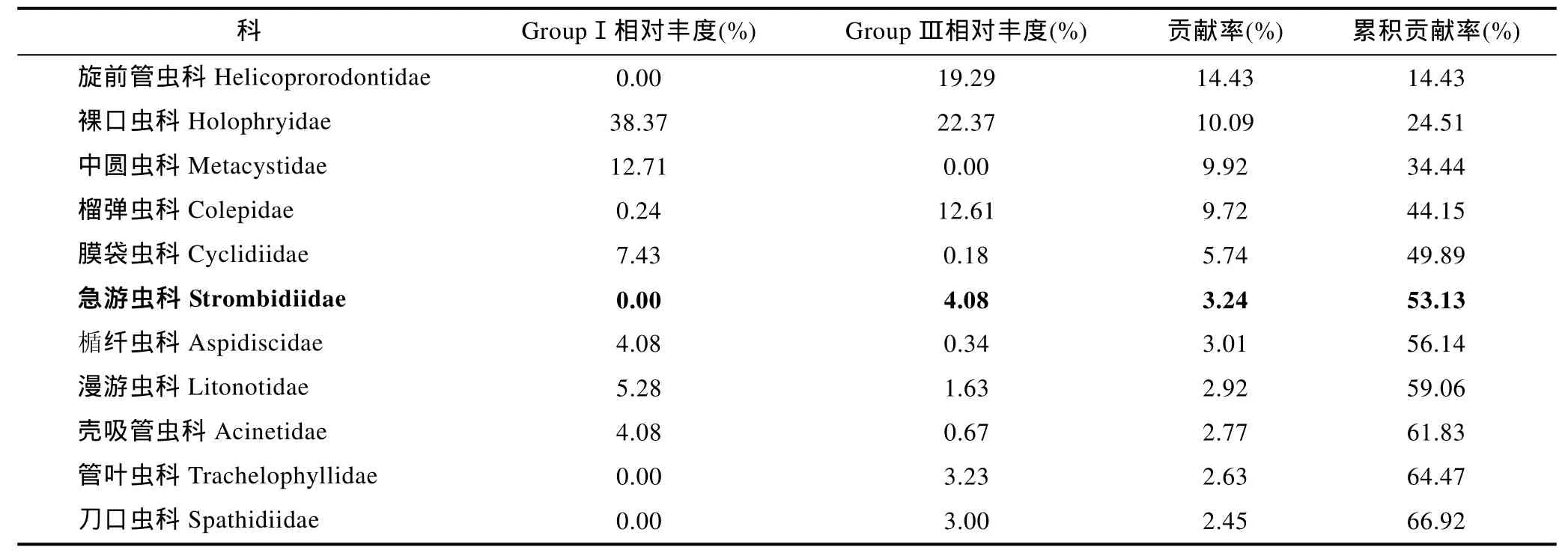
表6 基于科级水平的相对丰度聚类获得的形态检测组(Group Ⅰ)和cDNA测序组(Group Ⅲ)的非相似性贡献(粗体示浮游类群)Tab.6 Dissimilarity contribution of the ciliates species relate abundance at the level of family between Groups Ⅰ and Ⅲ (pelagic ciliates are indicated in boldface)
3 讨论
3.1 形态观察、DNA及其cDNA测序对海洋底栖纤毛虫多样性的评估效能
相较于传统的形态学方法, 高通量测序技术揭示了更高的真核生物多样性。Santoferrara等(2014)结合形态观察和焦磷酸测序评估浮游纤毛虫的多样性发现, 通过测序获得的多样性较形态学方法高一个数量级。但分子揭示的高多样性是真实存在, 还是一种假象, 或者是其他因素造成的, 成为一个广泛关注的问题。
本研究首次结合形态观察、DNA及其cDNA测序三种方法, 比较评估了黄海沉积物中纤毛虫的多样性。研究显示, 两种分子手段的结果更为相似, 形态学方法检获的物种多样性分别为DNA和cDNA测序方法检获的OTU数的1/2和1/3。分析认为, 这些差异的产生是由多种因素造成的。首先, 基于形态分类学的方法局限可导致物种多样性的低估。本研究采用的Ludox-QPS法(密度梯度离心结合定量蛋白银染色), 可在属/种水平上揭示大部分底栖纤毛虫的多样性 (Xuet al, 2010)。然而, 由于纤毛虫的固定及染色效能局限, 某些纤毛虫如核残迹纲的许多类群可能并未完整保存下来, 部分类群仅能鉴定到科甚至目级, 从而导致物种多样性不同程度的低估。
在DNA测序检获的76个属、174个OTUs中, 包含了浮游纤毛虫的 15个属、35个 OTUs。然而, 这些浮游类群的活动虫体在沉积物中是几乎不存在的,在底栖纤毛虫的形态学检测中鲜少出现。因此, 将这些类群考虑在内, 无疑会高估底栖纤毛虫的多样性。导致沉积物中大量浮游纤毛虫出现的原因在于, 沉积物不仅包括活动虫体的DNA, 同时包括一些裂解、释放的DNA及浮游类群的包囊DNA等(Torti et al,2015)。Dell'Anno等(2004)对亚得里亚海沿岸沉积物中胞外DNA降解速率的研究发现, DNA的周转时间由29到93天不等。因此, DNA方法检获的不仅有活动虫体, 还包括埋藏在沉积物中的历史DNA。
在cDNA测序检获的99个属、284个OTUs中, 18个属、37个OTUs为浮游类纤毛虫。这一结果表明, 沉积物中依然存在浮游纤毛虫的 RNA, 而通常认为RNA在生物死亡后会很快降解。Novitsky等(1986)对海洋沉积物中死亡微型生物 RNA的研究发现, 尽管RNA的降解速率远高于DNA的降解速率, 但14天后依然有30%—40%的RNA未降解。即使将浮游类群排除在外, cDNA方法相较DNA方法检获了更高的底栖纤毛虫多样性, 这可能主要是两种分子方法采用的样品量不同所致。样品量是影响微型生物多样性评价的重要因素; Penton等(2016)和Dolan等(2011)针对土壤和水体样品研究发现检获的物种数随样品量增加而增多。cDNA方法所用的沉积物样品量是DNA方法的2倍, 因此检获了更高的多样性。然而,形态学方法所用的沉积物样品量约为DNA测序样品量的3倍, 是cDNA测序样品量的1.5倍, 但所获多样性仍然较分子方法低, 这也间接表明分子手段较形态学方法而言在检获纤毛虫多样性上优势明显,可更全面地揭示沉积物中的纤毛虫多样性。
此外, 与形态学方法相比, 分子手段在鉴定稀有类群方面优势明显, 在仅通过分子手段检获的 84个属中, 有64个属相对丰度低于1%。在海洋沉积物中,大部分底栖纤毛虫以较低的丰度存在, 因此通过形态学检获不易。本研究通过形态学分析的6g沉积物中, 很大一部分纤毛虫仅发现一个个体。
3.2 三种方法检测的海洋底栖纤毛虫丰度/相对丰度比较
本研究使用 Ludox-QPS方法检测的结果表明,以裸口虫科及中圆虫科为主的前口纲纤毛虫丰度最高, 占了总丰度的 53%。这与课题组先前的研究结果是一致的。Meng等(2012)对 2007年7月黄海 48个站位的底栖纤毛虫研究发现, 前口纲的丰度可达总丰度的 45%; 周百灵等(2016)和 Zhou等(2016)分别对2010年和2011年7月和11月黄海底栖纤毛虫的研究发现, 前口纲的丰度所占比例分别为 40%—45%。
相较于此, DNA高通量测序揭示了更多的浮游纤毛虫类群, 其序列相对丰度竟达 90%。Zhao等(2016)在黄海陆架区沉积物中纤毛虫 DNA高通量测序分析显示, 浮游纤毛虫的相对丰度更达95%。可见,以沉积环境的总 DNA进行纤毛虫多样性研究, 不仅可以反映活动虫体的多样性信息, 同时还可反映“历史”群落以及包囊的多样性信息。
cDNA高通量测序检测的序列相对丰度结果与形态学方法类似, 底栖纤毛虫序列相对丰度占比约93%。其中, 形态学检测的个体丰度最高的裸口虫科在内的前口纲, 其序列相对丰度亦最高, 所占比例达35%。然而, 分子方法检测的对裸口虫科主要贡献的属是寄生的隐核虫属而非底栖生的裸口虫属, 推测由于NCBI中没有裸口虫属相关序列, 因此只能比对到相似度较高的隐核虫属所致。
值得注意的是, 测序所得的序列相对丰度无法反映环境中相应纤毛虫确切的丰度信息。Gong等(2013)通过单细胞定量 PCR对寡毛类及缘毛类纤毛虫核糖体 RNA基因拷贝数研究中发现, 相较其它原生生物和真菌, 部分纤毛虫的rDNA具有较高的拷贝数, 且其数量无论是在种内还是在种间均具有一定差异。因此, rDNA高通量测序所获各类群的序列相对丰度与群落中实际的个体相对丰度并无一一对应关系。尽管如此, 已有的研究还是通过序列的相对丰度来区分相对优势类群与稀有类群, 进行大致的界定。新近的研究也将序列的相对丰度与形态学方法所获的相对丰度做比较分析, 探索进行相对优势类群的比较(Bachy et al, 2013; Santoferrara et al, 2014)。本研究以相同方法对DNA及其cDNA测序结果进行了处理, 发现 DNA测序获得的浮游类纤毛虫序列所占比例高达 90%, 而 cDNA测序为 7%, 且相对丰度最高的类群与形态学方法相同。因此, 分析认为尽管纤毛虫存在DNA拷贝数不同的问题, 但较之DNA高通量测序, cDNA高通量测序检获的底栖纤毛虫在群落结构上与形态学方法更为接近。
4 结论
对比分析了基于形态学、DNA及其cDNA高通量测序检获的黄海沉积物中的纤毛虫多样性, 发现三种方法均受到多种因素的影响, 导致评价结果差异明显。分子手段有助于更全面地研究海洋沉积物中的纤毛虫多样性, 检获的多样性基本涵盖了形态学手段检获的所有底栖类群, 此外还包括了数量较多的浮游类群。相较DNA高通量测序, cDNA测序与形态学方法获得的结果更为一致, 在研究活动虫体多样性方面更有优势, 而 DNA测序方法可同时揭示包囊及过去群落信息。
致谢 本课题组孟昭翠、陈旭淼、李菊博士在Ludox-QPS方法及分类鉴定中给予协助, 詹子锋博士、维妙博士生协助采样, 在此谨致谢忱。
代仁海, 2012. 黄东海底栖纤毛虫多样性及微型底栖生物群落结构特点. 青岛: 中国科学院海洋研究所博士学位论文,1—93
宋微波, 沃 伦 A, 胡晓钟, 2009. 中国黄渤海的自由生纤毛虫. 北京: 科学出版社, 1—515
陈旭淼, 徐奎栋, 2014. 深海原生动物多样性研究进展. 海洋科学, 38(10): 119—126
周百灵, 维 妙, 李 菊等, 2016. 黄海底栖纤毛虫的群落结构与时空变化. 海洋与湖沼, 47(2): 336—345
徐奎栋, 2011. 海洋微型底栖生物的多样性与地理分布. 生物多样性, 19(6): 661—675
Bachy C, Dolan J R, López-García P et al, 2013. Accuracy of protist diversity assessments: morphology compared with cloning and direct pyrosequencing of 18S rRNA genes and ITS regions using the conspicuous tintinnid ciliates as a case study. The ISME Journal, 7(2): 244—255
Caporaso J G, Kuczynski J, Stombaugh J et al, 2010. QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7(5): 335—336
Carey P G, 1992. Marine Interstitial Ciliates: An Illustrated Key.London, New York, Tokyo, Melbourne, Madras: Chapman &Hall Identification Guide, 1—351
Dell'Anno A, Corinaldesi C, 2004. Degradation and turnover of extracellular DNA in marine sediments: ecological and methodological considerations. Applied and Environmental Microbiology, 70(7): 4384—4386
Doherty M, Tamura M, Vriezen J A C et al, 2010. Diversity of Oligotrichia and Choreotrichia ciliates in coastal marine sediments and in overlying plankton. Applied and Environmental Microbiology, 76(12): 3924—3935
Dolan J R, Stoeck T, 2011. Repeated sampling reveals differential variability in measures of species richness and community composition in planktonic protists. Environmental Microbiology Reports, 3(6): 661—666
Edgar R C, 2013. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10(10): 996—998
Edgar R C, Haas B J, Clemente J C et al, 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics,27(16): 2194—2200
Gong J, Dong J, Liu X H et al, 2013. Extremely high copy numbers and polymorphisms of the rDNA operon estimated from single cell analysis of oligotrich and peritrich ciliates. Protist, 164(3):369—379
Gong J, Shi F, Ma B et al, 2015. Depth shapes α- and β-diversities of microbial eukaryotes in surficial sediments of coastal ecosystems. Environmental Microbiology, 17(10): 3722—3737 Grattepanche J D, Santoferrara L F, Andrade J et al, 2014.Distribution and diversity of oligotrich and choreotrich ciliates assessed by morphology and DGGE in temperate coastal waters.Aquatic Microbial Ecology, 71(3): 211—221
Haas B J, Gevers D, Earl A M et al, 2011. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Research, 21(3):494—504
Hausmann K, Hülsmann N, Polianski I et al, 2002. Composition of benthic protozoan communities along a depth transect in the eastern Mediterranean Sea. Deep Sea Research Part I:Oceanographic Research Papers, 49(11): 1959—1970
Karayanni H, Christaki U, Van Wambeke F et al, 2004. Evaluation of double formalin—Lugol’s fixation in assessing number and biomass of ciliates: an example of estimations at mesoscale in NE Atlantic. Journal of Microbiological Methods, 56(3):349—358
Kim Y O, Ha S, Taniguchi A, 2008. Morphology and in situ sedimentation of the cysts of a planktonic oligotrich ciliate,Strombidium capitatum. Aquatic Microbial Ecology, 53(2):173—179
Kunin V, Engelbrektson A, Ochman H et al, 2010. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environmental Microbiology,12(1): 118—123
Lara E, Berney C, Harms H et al, 2007. Cultivation-independent analysis reveals a shift in ciliate 18S rRNA gene diversity in a polycyclic aromatic hydrocarbon-polluted soil. FEMS Microbiology Ecology, 62(3): 365—373
Lynn D H, 2008. The Ciliated Protozoa: Characterization,Classification, and Guide to the Literature. 3rd ed. Dordrecht,Netherlands: Springer
Lynn D H, Small E B, 2002. Phylum ciliophora. In: Lee J J,Leedale G F, Bradbury P C eds. An Illustrated Guide to the Protozoa. 2nd ed. Lawrence, Kansas, USA: Society of Protozoologists, Allen Press, 371—656
Magoč T, Salzberg S L, 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics,27(21): 2957—2963
Meng Z C, Xu K D, Dai R H et al, 2012. Ciliate community structure, diversity and trophic role in offshore sediments from the Yellow Sea. European Journal of Protistology,48(1): 73—84
Novitsky J A, 1986. Degradation of dead microbial biomass in a marine sediment. Applied and Environmental Microbiology,52(3): 504—509
Pawlowski J, Christen R, Lecroq B et al, 2011. Eukaryotic richness in the abyss: insights from pyrotag sequencing.PLoS One, 6(4): e18169
Penton C R, Gupta V V S R, Yu J L et al, 2016. Size matters:assessing optimum soil sample size for fungal and bacterial community structure analyses using high throughput sequencing of rRNA gene amplicons. Frontiers in Microbiology, 7: 824
Reid P P C, John A W G, 1978. Tintinnid cysts. Journal of the Marine Biological Association of the United Kingdom, 58(3):551—557
Rubino F, Belmonte G, Miglietta A M et al, 2000. Resting stages of plankton in recent North Adriatic sediments. Marine Ecology, 21(3—4): 263—284
Santoferrara L F, Grattepanche J D, Katz L A et al, 2014.Pyrosequencing for assessing diversity of eukaryotic microbes: analysis of data on marine planktonic ciliates and comparison with traditional methods. Environmental Microbiology, 16(9): 2752—2763
Stock A, Edgcomb V, Orsi W et al, 2013. Evidence for isolated evolution of deep-sea ciliate communities through geological separation and environmental selection. BMC Microbiology, 13: 150
Stoeck T, Bass D, Nebel M et al, 2010. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water.Molecular Ecology, 19(S1): 21—31
Torti A, Lever M A, Jørgensen B B, 2015. Origin, dynamics, and implications of extracellular DNA pools in marine sediments.Marine Genomics, 24: 185—196
Xu K D, Du Y F, Lei Y L et al, 2010. A practical method of Ludox density gradient centrifugation combined with protargol staining for extracting and estimating ciliates in marine sediments. European Journal of Protistology, 46(4):263—270
Zhao F, Xu K D, 2016. Molecular diversity and distribution pattern of ciliates in sediments from deep-sea hydrothermal vents in the Okinawa Trough and adjacent sea areas. Deep Sea Research Part I: Oceanographic Research Papers, 116:22—32
Zhou B L, Xu K D, 2016. Spatiotemporal variation in community structure of marine benthic ciliates in the Yellow Sea during and after macroalgal and giant jellyfish blooms. Chinese Journal of Oceanology and Limnology, 34(4): 629—641
