MoS2/TiO2复合催化剂的制备及其在紫外光下的光催化制氢活性
2017-11-01吴志娇刘建军朴玲钰
张 驰 吴志娇 刘建军 朴玲钰,*
(1北京化工大学化工资源有效利用国家重点实验室,北京 100029;2中科院标准与检测重点实验室,中科院纳米科学卓越创新中心,国家纳米科学中心,北京 100190)
MoS2/TiO2复合催化剂的制备及其在紫外光下的光催化制氢活性
张 驰1,2吴志娇2刘建军1,*朴玲钰2,*
(1北京化工大学化工资源有效利用国家重点实验室,北京 100029;2中科院标准与检测重点实验室,中科院纳米科学卓越创新中心,国家纳米科学中心,北京 100190)
为了研究复合光催化剂在光催化中的制氢效率,采用水热法制备了MoS2纳米片,然后通过水热法在MoS2纳米片上负载了TiO2纳米颗粒,形成了MoS2/TiO2异质结复合催化剂。采用冷场发射扫描电子显微镜(FE-SEM)、透射电子显微镜(TEM)、X射线衍射(XRD)、紫外-可见吸收光谱(UV-Vis)、拉曼光谱(Raman),X射线光电子能谱(XPS)对材料的结构和光学性能表征并进行分析。通过光催化制氢测试对光催化剂进行评价,实验结果表明,在波长为365 nm的紫外光照射下,最高光催化制氢速率为1004 μmol·h−1·g−1,对应的催化剂的MoS2含量为30%,其催化速率远大于单一的MoS2和TiO2,表明MoS2/TiO2复合催化剂在紫外光照下能显著提高光催化产氢性能。基于MoS2/TiO2复合光催化剂优越的光催化产氢性能,本文对复合光催化剂的产氢机理做了研究和分析。
二氧化钛;二硫化钼;异质结;复合结构;光催化制氢
1 引 言
对不可再生资源的过分依赖严重制约了我国经济社会的可持续发展。氢能作为一种清洁的理想的可再生能源,具有热值高、来源广、密度小、无污染、易贮存和运输等优点1,2。随着光催化技术的发展和成熟,利用半导体光催化剂分解水产氢成为有效解决能源短缺的方法之一。
TiO2是一种常见的半导体光催化剂,具有成本低、环境友好、化学稳定性高等优点。由于锐钛矿型TiO2带隙较宽(3.2 eV),只能吸收占太阳光约4%的紫外光,光能利用率较低。另外,由于大量的光生电子和空穴极易复合,减少了能够发生氧化还原反应的活性物种,使得TiO2光催化效率不高3−5。为了提高光催化剂的光催化制氢效率,贵金属掺杂通常能促进光催化产氢,然而由于高成本和对环境的不友好使得贵金属作为助催化剂并不是一种理想的选择。
二硫化钼(MoS2)作为一种二维层状的过渡金属硫化物,属六方晶系,由单层或多层二硫化钼组成具有“三明治夹心”层状结构6。在催化剂、储氢、锂电池、和光电子器件等方面有重要的应用7,8。据文献报道,在TiO2的表面生长 MoS2形成的复合新型催化剂,都能明显提高光催化效率。如Yuan等9通过水热法在 TiO2暴露{001}晶面上生长0.5% (w,质量分数) MoS2,光催化产氢速率达 2145 μmol·h−1·g−1,He 等10通过气相沉积法在TiO2纳米棒上竖直生长3% (w)的MoS2,光催化产氢速率达 4300 μmol·h−1·g−1,Zhou 等11通过水热法在TiO2纳米带上包围50% (w)的MoS2,光催化产氢速率达 1600 μmol·h−1·g−1。然而在 MoS2的纳米片上生长TiO2纳米颗粒形成的MoS2/TiO2新型复合催化剂,在紫外光下催化制氢仍然很少有文献报道。
本文首先采用水热法制备 MoS2纳米片,然后将 TiO2纳米粒子负载于 MoS2纳米片上。采用扫描显微镜(SEM)、透射显微镜(TEM)、X射线衍射(XRD)、紫外可见漫反射光谱(UV-Vis)、拉曼光谱、X射线光电子能谱(XPS)对材料进行了表征,并在紫外光照下,通过光催化制氢评价,讨论和分析了光催化剂制氢机理。
2 实验部分
2.1 实验试剂和仪器
本实验采用的化学试剂有:四水合钼酸钠(Na2MoO4·2H2O,分析纯)、硫脲(C2H5NS,分析纯)、氢氟酸(HF,分析纯40% (w))均由国药集团化学试剂有限公司生产,钛酸四丁酯(TBOT,分析纯)由北京兴津化工厂生产,无水乙醇(C2H5OH,分析纯)由北京市化学试剂厂生产,去离子水(H2O,高纯(18 MΩ·cm))。
本实验使用的仪器有:D8 Focus型X射线粉末衍射仪(XRD,德国布鲁克公司),Cu Kα辐射源,λ = 0.15406 nm;S-4800扫描电子显微镜(SEM,日本日立公司),扫描电压5 kV;T20透射电子显微镜(TEM,美国 FEI公司),加速电压 200 KV;Renishaw拉曼光谱分析仪(Renishaw,英国雷尼绍公司),激发波长514 nm;U-3900型UV-Vis吸收光谱(Hitachi U-3900);Escalab 250Xi X射线光电子能谱仪(XPS,美国赛默飞公司);气相色谱仪(TechcoMp GC 7900,上海天美科学仪器有限公司)。
2.2 催化剂的制备
2.2.1 MoS2的制备
准确称量 1 mmol四水合钼酸钠(1.235 g)和30 mmol硫脲(2.283 g)溶解在35 mL去离子水中不断搅拌形成透明溶液,转移至 100 mL 聚四氟乙烯乙烯内衬不锈钢高压反应釜中,将反应釜放置在鼓风干燥箱中220 °C反应18 h,反应结束后反应釜在鼓风干燥箱中冷却至室温,取出反应釜,收集反应釜内杯中的产物分别用去离子水和乙醇洗涤数次,放置在鼓风干燥箱60 °C干燥12 h。干燥完毕研磨均匀,保存在密封袋中待用。
2.2.2 MoS2/TiO2复合催化剂的制备
MoS2/TiO2(30% (w) of MoS2)催化剂制备:准确称量0.1 g MoS2黑色粉末分散在含有5 mL乙醇的容量为20 mL的聚四氟乙烯内杯中不断搅拌,超声10 min,往分散液中缓慢滴加1.0 mL钛酸四丁酯,并不断搅拌,滴加完毕后搅拌30 min,再加入0.12 mL HF,搅拌2 h,把反应釜转移至鼓风干燥箱中180 °C 保持12 h,反应结束后反应釜在鼓风干燥箱中自然冷却至室温,取出反应釜,收集反应釜内杯中的产物,分别用去离子水和无水乙醇洗涤几次,放置鼓风干燥箱60 °C 干燥12 h。干燥完毕后研磨均匀,保存在密封袋中待用。
2.3 光催化制氢实验
光催化制氢实验的光源是300 W氙灯,波长365 nm,甲醇作为牺牲剂,光催化反应器距离光源3 cm。具体操作为:在50 mL的石英管里加入20 mg光催化剂,再加入10 mL体积分数为20%的甲醇水溶液,往石英管里通入N230 min,排除石英管里的氧气,用橡胶塞和密封胶带密封石英管口,放置于300 W氙灯光源下照射,石英管里的气体组分通过气相色谱仪(TechcoMp GC 7900)检测。
3 结果与讨论
3.1 催化剂的XRD和拉曼分析
所制备样品的XRD如图1所示,纯TiO2的XRD衍射峰与锐钛矿 TiO2的 PDF卡片(JPCDS card No. 21-1272)一致,表明制备的TiO2为锐钛矿型,位于2θ等于25.1°的衍射峰对应锐钛矿 TiO2的(101)晶面。纯MoS2的衍射峰分别位于14.00°、33.28°、39.44°、58.66°,归属于MoS2的(002)、(100)、(103)、(110)晶面,与六方相MoS2的PDF卡片(a =b = 0.316 nm,c = 1.230 nm,JPCDS cards No.37-1492)相匹配12,13。由于MoS2的(002)衍射峰反应了 MoS2的堆垛程度和结晶度,(002)衍射峰强度越大,表明MoS2具有较高的结晶度和较好的堆垛层结构14,15。MoS2/TiO2复合材料的 XRD谱图中,MoS2和TiO2的主要衍射峰都能观察到,MoS2在 2θ等于14.00°的衍射峰对应于 MoS2沿 c轴方向的层间距为0.62 nm。随着MoS2的比例增加,对应的MoS2的特征衍射峰强度增加,相应的TiO2的特征衍射峰强度减小。
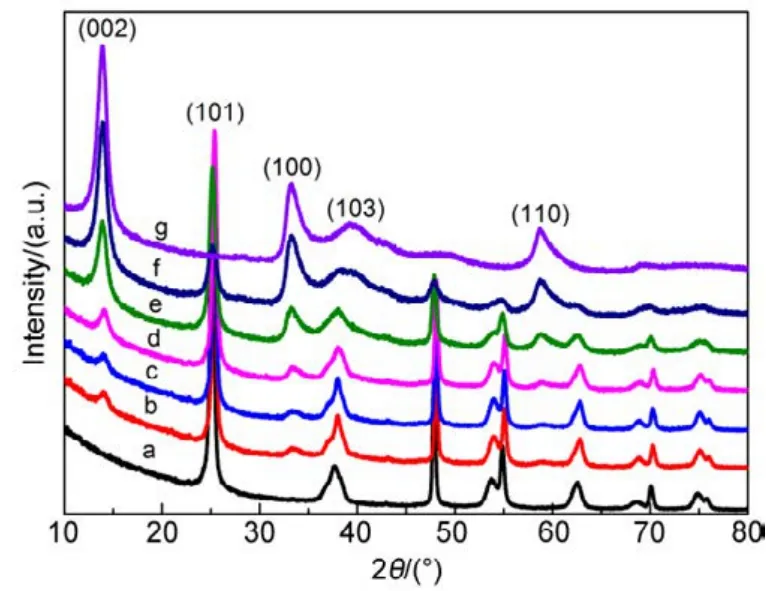
图1 所制备样品的XRD谱图Fig.1 XRD patterns of as-prepared samples.
制备样品的拉曼光谱见图 2。纯 TiO2的拉曼散射峰位于 144 cm−1(E1g)、394 cm−1(B1g)、514 cm−1(A1g)、636 cm−1(E2g),对应锐钛矿 TiO2的特征峰。在 385、409、453 cm−1出现 MoS2的 E2g、A1g、E1u特征振动峰16,17,其中E2g与A1g的波数差为24 cm−1,表明MoS2为多层结构18。在MoS2/TiO2复合结构的拉曼光谱中,E2g和A1g的振动峰位置分别在 381和 407 cm−1,和纯 MoS2的 E2g和 A1g比较,分别蓝移4和2 cm−1;同时,E1g的振动峰位置在145 cm−1处,和纯的TiO2的E1g比较,红移1 cm−1,MoS2/TiO2复合结构中拉曼特征峰发生位移,或许由于 TiO2晶体颗粒与 MoS2界面的相互作用,形成了化学键,使得二者界面张力变大,导致拉曼特征峰发生位移。因此这也证明在MoS2/TiO2复合结构的存在。
3.2 催化剂的SEM和TEM分析
图3(a)为纯MoS2的扫描图。纯MoS2是黑色粉末,是由二维纳米片组装成的三维分级结构,呈花瓣状。图3(b, c)为MoS2/TiO2(30%(w))复合结构。从图中可以看出,TiO2晶体颗粒生长在MoS2的纳米片上,与图 3(a)中 MoS2相比,MoS2/TiO2复合材料的形貌略有变化,MoS2纳米片更加舒展,结构疏松。图3(d)为高分辨率下的MoS2/TiO2(30% (w))复合材料扫描图,可以看出,TiO2纳米颗粒紧密负载在MoS2纳米片上,颗粒之间没有团聚,粒径平均约15 nm。
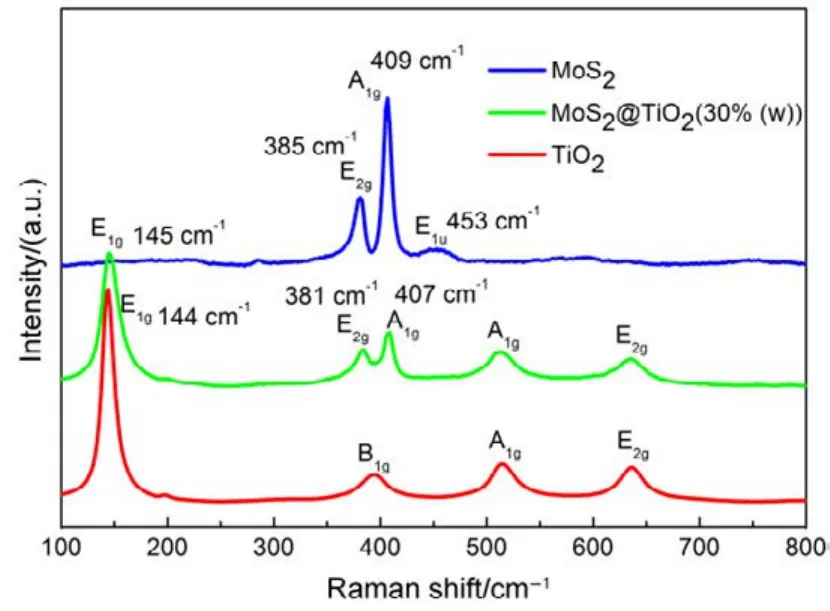
图2 所制备样品的Raman光谱Fig.2 Raman spectra of as-prepared samples.

图3 所制备样品的扫描电镜照片Fig.3 SEM images of as-prepared samples.

图4 所制备样品的透射电镜照片Fig.4 TEM images of as-prepared samples.
图4(a)为纯MoS2透射电镜图,从图中可以看出,纯的MoS2为薄纳米片组装而成,呈“絮状结构,片层薄,边缘卷曲。图 4(b, c)为所制备的样品复合结构,从图可以看出,粒径约为15 nm的TiO2纳米颗粒不仅负载在 MoS2的平面内部,在MoS2的活性边缘也有负载。图4(d)为高分辨率下的透射电镜图,从图中可以观察到,MoS2层间距为0.62 nm,对应六方相MoS2的(002)晶面,TiO2晶格条纹0.235 nm对应(001)晶面,表明在MoS2上负载的TiO2晶体颗粒暴露高能面{001}晶面。同时也可以看到MoS2平面的晶格条纹是不连续的,表明存在缺陷位点,从而增加MoS2的活性部位。
3.3 催化剂的紫外-可见吸收光谱分析
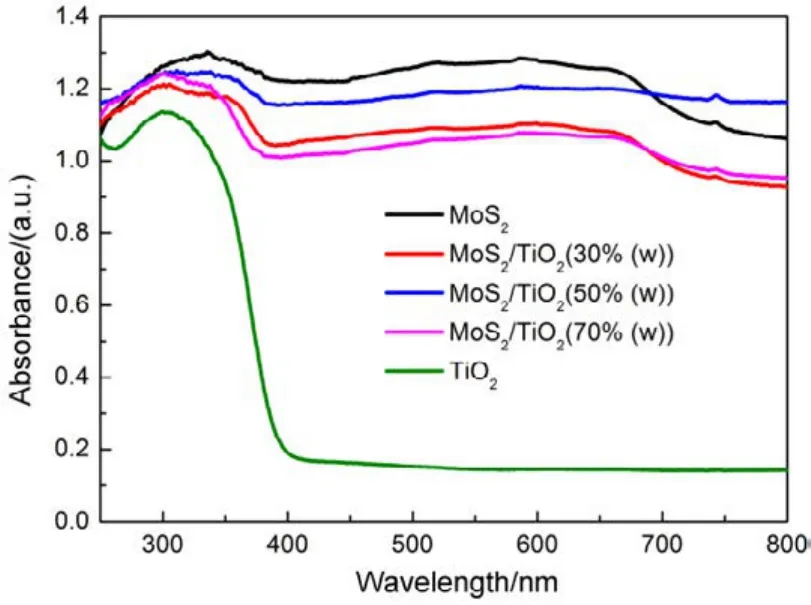
图5 制备样品的紫外-可见吸收光谱Fig.5 UV-Vis spectra of as-prepared sample.
不同样品的紫外-可见漫反射图谱如图 5所示,纯TiO2的吸收边缘在389 nm,对应的带隙为3.19 eV,MoS2的吸收边缘在939 nm,对应的带隙为1.32 eV19。禁带宽度决定了TiO2只能被小于389nm的紫外光激发,而对大于389 nm的可见光只有散射和反射作用。而纯MoS2为黑色粉末,带隙小,能吸收波长范围在300−800 nm的光,同时伴随着热能释放20。和纯 TiO2相比,MoS2/TiO2复合材料的吸收强度明显增加,吸收边红移,复合材料对紫外光吸收强度从大到小分别为 50%(w)、70% (w)、30% (w) MoS2/TiO2,这些现象可能是由于 MoS2和 TiO2在界面形成化学键21,改变了MoS2和TiO2的界面状态。
3.4 MoS2/TiO2复合催化剂的XPS分析
MoS2/TiO2(30% (w))的XPS光谱如图6所示,从图 6(a)中可以看出,样品 MoS2/TiO2(30% (w))含有Ti、O、Mo、S四种元素。从图6(b, c)看到Ti 2p3/2、Ti 2p1/2和O 1s的结合能分别为458.9、464.6 和 530.1 eV,表明 Ti4+氧化态10,22。从图 6(d,e)可以看出,在样品 MoS2/TiO2(30% (w))中,Mo 3d5/2、Mo 3d3/2的结合能分别为228.7、231.8 eV,对应MoS2中的Mo4+,结合能分别在161.5和162.7 eV的吸收峰对应MoS2中的S 2p3/2和S 2p1/2。同时,结合能为225.8 eV的吸收峰存在,表明样品MoS2/TiO2中 Mo6+存在,这是由于 MoS2中部分Mo4+被氧化11,23。有文献报道24纯 MoS2中 Mo 3d5/2、Mo 3d3/2、S 2p3/2和S 2p1/2的结合能分别为229.3、232.5、162.3和163.3 eV,表明随着TiO2纳米颗粒生长在MoS2片状结构上,S和Mo的结合能都降低,进一步证明 MoS2与 TiO2之间产生了化学键,表明 TiO2纳米颗粒成功引入 MoS2片状结构上。
3.5 MoS2/TiO2复合催化剂的光催化制氢活性
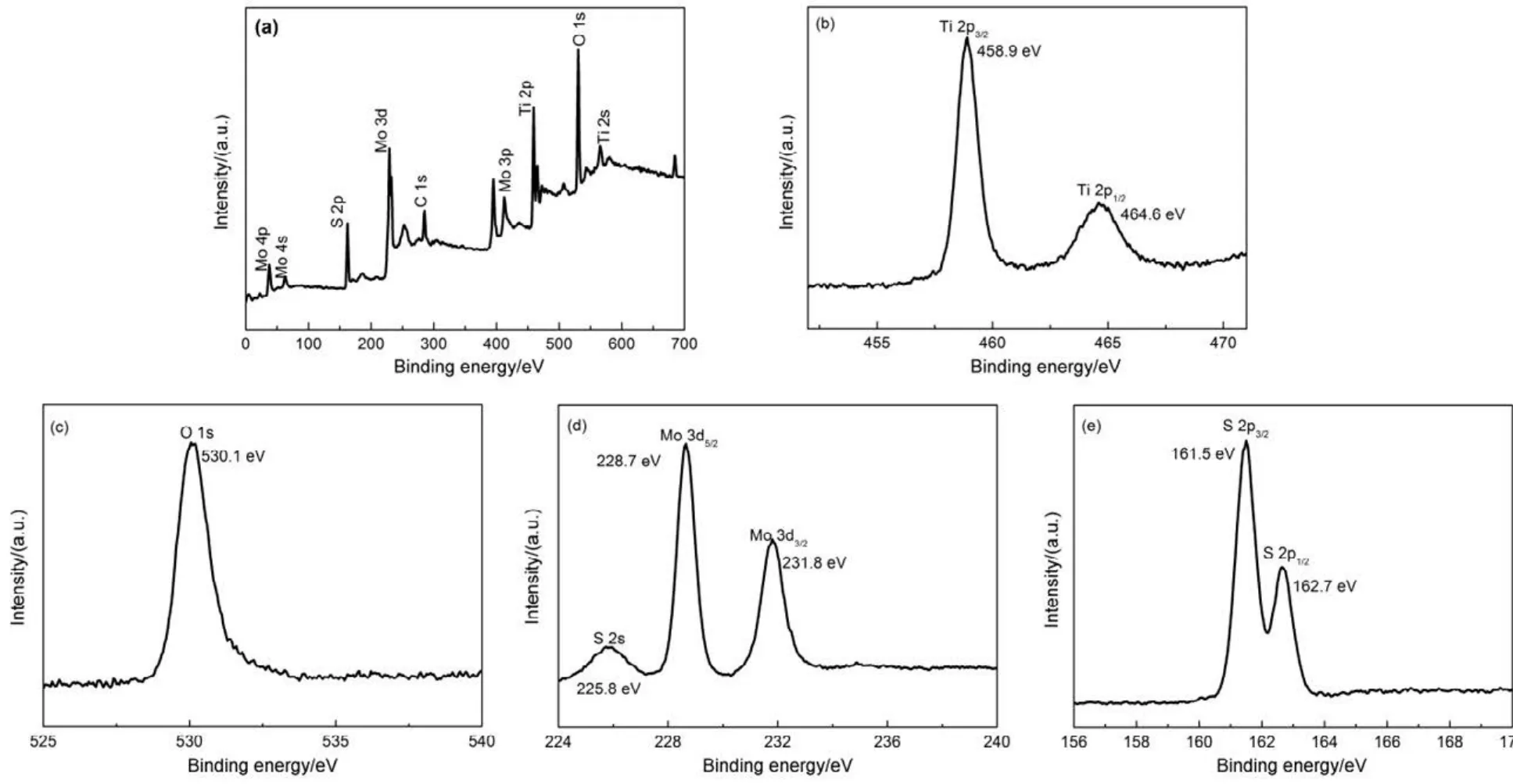
图6 MoS2/TiO2(30% (w))的XPS光谱Fig.6 XPS spectra of MoS2/TiO2(30% (w)).
图 7为不同催化剂的光催化产氢速率图,如图所示,纯 MoS2和纯 TiO2的产氢速率非常小,表现出很低的光催化产氢活性。由于所制备的MoS2片层较厚,带隙小,导带位置较低,决定其不具备有效制氢能力。而TiO2虽然导带位置较高,但是大多数产生的电子和空穴在迁移至表面之前复合,消弱了光催化能力。5%、10%、30%、50%、70% (w) MoS2/TiO2复合催化剂的产氢速率分别为130、134、1004、26、23 μmol·h−1·g−1,当载体MoS2的含量为30%时,复合光催化剂的产氢效率最高,这主要有三个方面的原因:一是在MoS2/TiO2复合结构中,TiO2是光生电子的主要提供者,足够含量的TiO2有利于提高更多迁移至表面的电子;二是由于纯MoS2的活性位点在边缘区域,而在平面区域呈现惰性10,25,本实验通过水热法 TiO2颗粒负载在 MoS2片层结构过程中,由于高温反应使得完整的MoS2表面缺陷增多,活性部位增加。三是MoS2比表面积较大,较高的比表面积有利于光催化剂对甲醇溶液的吸附作用,有利于提高光催化制氢效率。
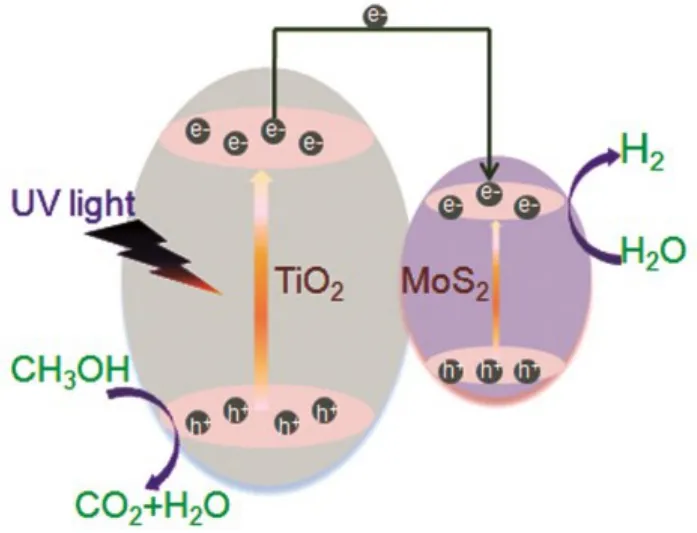
图8 光催化反应机理Fig.8 Mechanism of photocatalytic reaction.
3.6 复合催化剂MoS2/TiO2的光催化制氢机理讨论
复合光催化剂 MoS2/TiO2的制氢机理如图 8所示,在紫外光激发下,TiO2价带上电子跃迁至导带,同时价带(VB)上留下空穴。由于TiO2的导带(CB)电位比 MoS2的导带电位更负(ECB(TiO2) =−0.51 V,ECB(MoS2) = −0.16 V)9,因此 TiO2把电子传输至MoS2。不少文献报道MoS2活性边缘的活性S原子能提高光催化活性26−28,Yan等29证明MoS2的不饱和S原子能增加电催化活性,Xie等25证明 MoS2表面缺陷更多不饱和 S原子裸露在外,使得MoS2的活性部位增加。本实验中MoS2片层结构中,一部分 TiO2纳米颗粒在 MoS2的边缘活性部位,另一部分 TiO2颗粒在 MoS2平面结构上,由于MoS2表面缺陷也能形成活性部位,使得MoS2的活性中心接受电子,吸附甲醇水溶液中的H+,形成H230,这时MoS2的作用和Pt、Pd等贵金属的作用类似,但是MoS2有较大的比表面积与TiO2颗粒形成异质结,同时较大的比表面积有利于对甲醇水溶液的吸附。由于TiO2的价带底电位比甲醇氧化电位更正,能甲醇氧化为 CO2和H2O。因此MoS2/TiO2异质结复合催化剂能够显著提高光催化产氢活性。
4 结 论
本文采用水热法制备了MoS2/TiO2复合材料,对材料做了一系列表征,并在紫外光照射下进行材料的光催化制氢测试,分析复合材料的光催化产氢机理,得出以下三点结论:
(1) SEM和TEM照片表明在MoS2/TiO2复合结构中,TiO2纳米颗粒负载在MoS2片状结构上,拉曼光谱表明MoS2和TiO2产生作用力,XPS光谱表明复合材料由MoS2和TiO2构成,UV-Vis光谱表明 MoS2/TiO2复合材料在紫外光区域吸收边红移,XRD谱图表明,MoS2/TiO2复合材料晶型没有改变。
(2) 在紫外光照射下的光催化制氢测试表明,MoS2/TiO2复合材料能显著提高光催化制氢活性。其中MoS2含量为30%时,光催化制氢速率为1004 μmol·h−1·g−1。
(3) MoS2作为助催化剂在光催化制氢的作用和贵金属类似,但是MoS2具有较大的比表面积,独特的边缘活性部位,来源广,成本低,环境友好等优点,使得MoS2和半导体复合形成的体系在光催化Z中有更大的潜力。
(1) Balat, M. International Journal of Hydrogen Energy 2008, 33 (15),4013. doi: 10.1016/j.ijhydene.2008.05.047
(2) Zheng, J. Y.; Zhang, J. F.; Chen, L. X.; W, G.; Gu, C. H.; Zhao, Y.Z.; Meng, B. J. Saf. Environ. 2016, 16 (6), 144. [郑津洋, 张俊峰,陈霖新, 王 赓, 顾超华, 赵永志, 蒙 波. 安全与环境学报,2016, 16 (6), 144.] doi: 10.13637/j.issn.1009-6094.2016.06.029
(3) Bai, S.; Wang, L. M.; Chen, X. Y.; Du, J. T.; Xiong, Y. J. Nano Res.2015, 8 (1), 175. doi: 10.1007/s12274-014-0606-9
(4) Liu, Y. M.; Zhang, W. G.; Liang, W.; Wang, H. X.; Yu, B. Rare Metal Mat. Eng. 2015, 44 (7), 1754. [刘一鸣, 张王刚, 梁 伟, 王红霞, 余 彬. 稀有金属材料与工程, 2015, 44 (7), 1754.]
(5) Yang, J. H.; Wang, D. G.; Han, H. X.; Li, C. Acc. Chem. Res. 2013,46 (8), 1900. doi: 10.1021/ar300227e
(6) Gu, P. C.; Zhang, K. L.; Feng, Y. L.; Wang, F.; Miao, Y. P.; Han, Y.M.; Zhang, H. X. Acta Phys. Sin. 2016, 65 (1), 18102. [顾品超, 张楷亮, 冯玉林, 王 芳, 苗银萍, 韩叶梅, 张韩霞. 物理学报,2016, 65 (1), 18102.] doi: 10.7498/aps.65.018102
(7) Wang, C. X.; Lin, H. H.; Liu, Z. Y.; Wu, J. P.; Xu, Z. Z.; Zhang, C.Part. Part. Syst. Charact. 2016, 33 (4), 221.doi: 10.1002/ppsc.201500222
(8) Wang, X.; Song, L.; Chen, L.; Song, H. H.; Zhang, Y. P. Adv. Mater.Chem. 2015, 2 (4), 49. [王 轩, 宋 礼, 陈 露, 宋欢欢, 张永平.材料化学进展, 2015, 2 (4), 49.] doi: 10.12677/AMC.2014.24008
(9) Yuan, Y. J.; Ye, Z. J.; Lu, H. W.; Hu, B.; Li, Y. H.; Chen, D. Q.;Zhong, J. S.; Yu, Z. T.; Zou, Z. G. ACS Catal. 2015, 6 (2), 532.doi: 10.1021/acscatal.5b02036
(10) He, H. Y.; Lin, J. H.; Fu, W.; Wang, X. L.; Wang, H.; Zeng, Q. S.;Gu, Q.; Li, Y. M.; Yan, C.; Tay, B. K.; Xue, C.; Hu, X.; Liu, Z. Adv.Energy Mater. 2016, 6 (14). doi: 10.1002/aenm.201600464
(11) Zhou, W. J.; Yin, Z. Y.; Du, Y. P.; Huang, X.; Zeng, Z. Y.; Fan, Z.X.; Liu, H.; Wang, J. Y.; Zhang, H. Small 2013, 9 (1), 140.doi: 10.1002/smll.201201161
(12) Wang, J.; Liu, J. L.; Chao, D. L.; Yan, J. X.; Lin, J. Y.; Shen, Z. X.Adv. Mater. 2014, 26 (42), 7162. doi: 10.1002/adma.201402728
(13) Li, Y. G.; Wang, H. L.; Xie, L. M.; Liang, Y. Y.; Hong, G. S.; Dai, H.Y. J. Am. Chem. Soc. 2011, 133 (19), 7296. doi: 10.1021/ja201269b
(14) Zhang, C, X.; Zhang, X. X.; Tao, H. J. Acta Phys. -Chim. Sin. 2014,30 (10), 1963. [张传香, 张晓雪, 陶海军. 物理化学学报, 2014, 30(10), 1963.] doi: 10.3866/PKU.WHXB201408043
(15) Tye, C. T.; Smith, K. J. Catal. Today 2006, 116 (4), 461.doi: 10.1016/j.cattod.2006.06.028
(16) Shen, M.; Yan, Z. P.; Yang, L.; Du, P. W.; Zhang, J. Y.; Xiang, B.Chem. Commun. 2014, 50 (97), 15447. doi: 10.1039/C4CC07351G
(17) Li, H. D.; Wang, Y.; Chen, G. H.; Sang, Y. H.; Jiang, H. D.; He, J. T.;Li, X.; Liu, H. Nanoscale 2016, 8 (11), 6101.doi: 10.1039/C5NR08796A
(18) Lin, T. Z.; Kang, B. T.; Jeon, M. H.; Huffman, C.; Jeon, J. H.; Lee, S.J.; Kim, K. N. ACS Appl. Mater. Interfaces 2015, 7 (29), 15892.doi: 10.1021/acsami.5b03491
(19) Ho, W. K.; Yu, J. C.; Lin, J.; Yu, J. G.; Li, P. S. Langmuir 2004, 20(14), 5865. doi: 10.1021/la049838g
(20) Li, D. F.; Zheng, J.; Chen, X. Y.; Zou, Z. G. Prog. Chem. 2007, 19(4), 464. [李敦钫, 郑 菁, 陈新益, 邹志刚. 化学进展, 2007, 19(4), 464.]
(21) Liu, H.; Lv, T.; Zhu, C. K.; Su, X.; Zhu, Z. F. J. Mol. Catal. A: Chem.2015, 396, 136. doi: 10.1016/j.molcata.2014.10.002
(22) Lee, Y. H.; Zhang, X. Q.; Zhang, W. J.; Chang, M. T.; Lin, C. T.;Chang K, D.; Li, L. J.; Lin, T. W. Adv. Mater. 2012, 24 (17), 2320.doi: 10.1002/adma.201104798
(23) Chen, Z.; Cummins, D.; Reinecke, B. N.; Clark, E.; Sunkara, M. K.;Jaramillo, T. F. Nano Lett. 2011, 11 (10), 4168.doi: 10.1021/nl2020476
(24) Wang, H. W.; Skeldon, P.; Thompson, G. E. Surf. Coat. Technol.1997, 91 (3), 200. doi: 10.1016/S0257-8972(96)03186-6
(25) Xie, J. F.; Zhang, H.; Li, S.; Wang. R. X.; Sun, X.; Zhou, M.; Zhou, J.F.; Xie, Y. Adv. Mater. 2013, 25 (40), 5807.doi: 10.1002/adma.201302685
(26) Karunadasa, H. I.; Montalvo, E.; Sun, Y. J.; Majda, M.; Long, J. R.Chang, C. J. Science 2012, 335 (6069), 698.doi: 10.1126/science.1215868
(27) Hinnemann, B.; Moses, P. G.; Bonde, J.; Jorgensen, K. P.; Nielsen, J H.; Horch, S.; Norskov, J. K. J. Am. Chem. Soc. 2005, 127, 5308.doi: 10.1021/ja0504690
(28) Chang, K.; Mei, Z. W.; Wang, T.; Kang, Q.; Ouyang, S. X.; Ye, J. H.ACS Nano 2014, 8 (7), 7078. doi: 10.1021/nn5019945
(29) Yan, Y.; Xia, B. Y.; Ge, X. M.; Liu, Z. L.; Wang, J. Y.; Wang, X.ACS Appl. Mater. Interfaces 2013, 5, 12794.doi: 10.1021/am404843b
(30) Wang, Y.; Li, G.; Li, P. W.; Hu, J.; Zhao, Q. H. Semicond Opt. 2016,No. 4, 461. [王 毅, 李 刚, 李朋伟, 胡 杰, 赵清华. 半导体光电, 2016, No. 4, 461.] doi: 10.16818/j.issn1001-5868.2016.04.002
Preparation of MoS2/TiO2Composite Catalyst and Its Photocatalytic Hydrogen Production Activity under UV Irradiation
ZHANG Chi1,2WU Zhi-Jiao2LIU Jian-Jun1,*PIAO Ling-Yu2,*
(1State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R.China;2CAS Key Laboratory of Standardization and Measurement for Nanotechnology, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, P. R. China)
To study the activity of a composite photocatalyst for photocatalytic hydrogen production, we prepared and loaded MoS2nanosheets with TiO2nanoparticles using a hydrothermal method, thus forming a MoS2/TiO2heterojunction composite catalyst. The structural and optical properties of the catalyst were characterized and analyzed by field-emission scanning electron microscopy, high-resolution transmission electron microscopy, X-ray powder diffraction, UV-Vis absorption spectra, Raman spectroscopy, and X-ray photoelectron spectroscopy. The activity of the photocatalyst was evaluated by its photocatalytic hydrogen production rate. The corresponding MoS2content of the catalyst was found to be 30%, and upon the exposure to 365nm UV light, a high photocatalytic hydrogen production rate of 1004 µmol−1·h−1·g−1was obtained. The catalytic rate is much greater than that obtained with MoS2or TiO2catalysts. The high hydrogen production rate indicated that the use of a MoS2/TiO2composite catalyst can significantly improve the UV-induced photocatalytic hydrogen production performance. Because of the excellent photocatalytic hydrogen production performance of the MoS2/TiO2composite, we studied andanalyzed the hydrogen production mechanism.
TiO2; MoS2; Composite structure; Heterojunction; Photocatalytic hydrogen production
January 16, 2017; Revised: April 11, 2017; Published online: April 14, 2017.
O649
10.3866/PKU.WHXB201704141 www.whxb.pku.edu.cn
*Corresponding authors. LIU Jian-Jun, Email: 13611012376@163.com; ljj-717@163.com. PIAO Ling-Yu, Email: piaoly@nanoctr.cn.The project was supported by the Ministry of Science and Technology of China (2016YFA0200900, 2016YFF0203803).
科技部重点研发计划项目(2016YFA0200900, 2016YFF0203803)资助
© Editorial office of Acta Physico-Chimica Sinica
