全外显子组测序对肥厚型心肌病猝死者的基因分析
2017-09-26许传超白云志许心舒吕国丽赖小平林汉光邝文健
许传超,白云志,许心舒,吕国丽,赖小平,陈 锐,林汉光,邝文健
(1.广东医科大学,广东 东莞523808;2.广东医科大学司法鉴定中心,广东 东莞523808;3.广州市公安局番禺区分局,广东 广州511400;4.广州市刑事科学技术研究所,广东 广州510030)
·论 著·
全外显子组测序对肥厚型心肌病猝死者的基因分析
许传超1,2,白云志3,许心舒4,吕国丽4,赖小平1,2,陈 锐1,2,林汉光1,2,邝文健1,2
(1.广东医科大学,广东 东莞523808;2.广东医科大学司法鉴定中心,广东 东莞523808;3.广州市公安局番禺区分局,广东 广州511400;4.广州市刑事科学技术研究所,广东 广州510030)
目的 在全外显子组水平分析1例肥厚型心肌病(hypertrophic cardiomyopathy,HCM)猝死病例的相关致病性基因突变。 方法 对1例具有HCM病理学特征的猝死病例样本,利用Illumina®HiSeq 2500平台进行全外显子组测序(whole exome sequencing,WES)。测序数据分析以hg19为参照序列,筛选可疑的单核苷酸变异位点,通过PhyloP、PolyPhen-2、SIFT等软件进行突变的保守性和功能分析。 结果 经过筛选,发现该病例的MYBPC3基因发生C719R杂合突变。 结论 利用二代测序技术进行全外显子组水平的分子解剖(基因突变检测和分析),有助于明确HCM的分子机制,并为死因分析提供新的方法和思路。
法医病理学;心肌病,肥厚性;猝死,心脏;外显子组;突变
肥厚型心肌病(hypertrophic cardiomyopathy,HCM)是常见的常染色体显性遗传病,发病率在1/500左右,是引起青壮年心源性猝死的主要心肌病[1]。其主要病理改变为心室肌非对称性肥厚、心室腔不扩张,心肌细胞肥大、排列紊乱,心肌纤维化和心肌缺血等。主要临床表现包括晕厥、心绞痛,心功能不全及猝死[2],60%~70%的HCM患者有家族史,称之为家族性HCM(familial HCM,FHCM),20%~30%为散发型HCM[1],HCM的首发症状即可为猝死。因此,及早进行该遗传性心肌病的诊断,有助于及早采取干预措施来控制HCM的疾病进展和预防猝死发生,对FHCM家庭及高风险人群有着重要意义。
自1989年首次在一FHCM家系中发现β-肌球蛋白重链基因(MYH7B)突变与HCM相关以来,目前研究发现至少14个致病基因,多达上千种突变体与HCM相关[3]。其中,MYH7B和肌球蛋白结合蛋白C基因(MYBPC3)是两个最主要的致病基因,近80%的FHCM与这些基因相关。散发性的HCM患者也与这些基因的突变相关。不过,HCM的临床表型存在着异质性,突变基因的基因型与表型关系不确定。因此,要确定新发现的HCM病例的致病突变类型,需全面筛查这10多个基因的全部序列。本研究利用全外显子组测序(whole exome sequencing,WES)技术对1例HCM猝死病例进行遗传学检测。
1 材料与方法
1.1 研究对象
猝死病例1例,女,48岁,农民,在田间工作时猝死。家属否认有家族心脏病史。
1.2 尸体解剖及取材
经系统尸体解剖检查,并取心脏、大脑、肝、肾、肺等主要器官行组织病理学检验。取心血进行常见毒(药)物检验。
1.3 全外显子组测序
1.3.1 基因组DNA提取
取尸体心血2 mL,利用全血基因组DNA提取试剂盒(DP1801,北京百泰克生物技术有限公司),提取基因组DNA。
1.3.2 外显子文库构建与测序
(1)使用Illumina TruSeq DNA样品制备试剂盒(美国Illumina公司)富集DNA序列,并将富集的DNA序列变性解链为单链DNA。
(2)加入生物素标记的DNA探针进行液相杂交,探针为外显子区域特异探针。
(3)在反应液中加入链霉亲和素磁珠,富集与探针杂交的目标序列。
(4)通过磁性吸附将与磁珠结合的DNA片段从溶液中脱离,富集到的DNA片段随后从磁珠上洗脱。经扩增后,外显子测序文库构建完成。
(5)对构建的外显子文库用Illumina®HiSeq 2500平台(美国Illumina公司)进行高通量测序。
1.4 生物信息学分析
用文献[4]方法,以hg19为参照序列,确定样本的单核苷酸变异位点(single nucleotide variant,SNV),筛选出等位基因频率<1%的SNV,进一步确定发生在猝死相关基因范围内的SNV。
利用Sanger法对筛选出的基因突变进行测序验证。
运用在线生物大分子功能预测软件PhyloP、PolyPhen-2(http://genetics.bwh.harvard.edu/pph2/)和SIFT(http://sift.jcvi.org/www/SIFT_chr_coords_submit. html)等对全外显子组测序结果中检测到的基因突变进行编码产物蛋白质的功能预测分析。
2 结 果
2.1 法医病理学检验
心脏质量550 g,左心室壁厚1.3 cm,右心室壁厚0.4 cm,室间隔厚1.8 cm(图1);室间隔厚度/左心室游离壁厚度=1.38;肺动脉瓣周径7.5 cm,二尖瓣周径7.9 cm,三尖瓣周径9.3 cm,主动脉瓣周径8.0 cm,主动脉根部管壁见粥样斑块,冠状动脉通畅。光镜下可见心肌细胞肥大(图2),部分心肌纤维呈波浪状排列,多处心肌陈旧性小梗死瘢痕形成,室间隔肌部及左心室壁见急性梗死灶伴少量淋巴细胞浸润。全身其他主要器官未发现明显致死性病理改变。常见毒(药)物检验阴性。
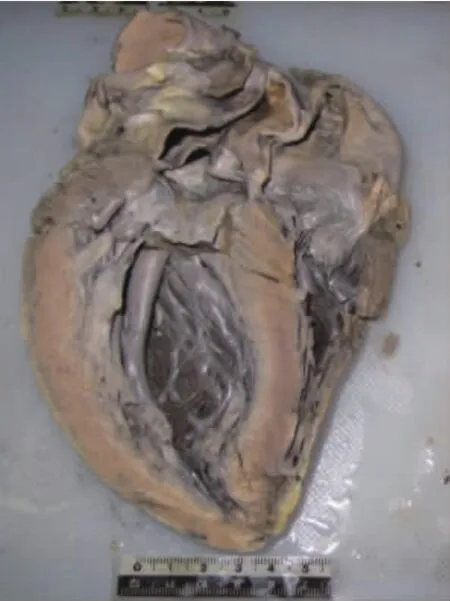
图1 心脏冠状面后面观
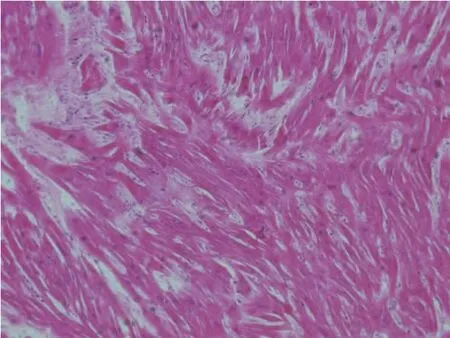
图2 左心室心肌HE×200
2.2 全外显子组测序及生物信息学分析结果
经过与hg19标准参考序列比对及与dbSNP等数据库信息比对,得到样本全外显子组及部分侧翼序列内的单核苷酸多态性(single nucleotide polymorphism,SNP)信息(表1);通过PhyloP、PolyPhen-2、SIFT等软件分析,得到4个疑似致病性基因突变位点(表2)。

表1 全外显子组测序结果与数据库外显子及临近侧翼区域参考序列的比对结果

表2 样本在心肌细胞结构及功能相关基因的外显子区域发现的部分SNV(错义突变,等位基因频率<1%)
2.3 Sanger法测序验证
利用“外显子区域错义突变(missense)-心肌细胞表达基因-基因频率<1%”的筛选。首先从20 644个外显子区域的SNV(包括SNP)中,筛选出分布在心肌细胞功能相关基因范围内的1 362个,其中,分布于外显子区域、基因频率<1%的非同义SNV共9个。通过比对dbSNP数据库相关SNV(包括SNP)位点的信息,以及SIFT和PolyPhen-2等软件的预测分析,最终筛选出可疑致病基因突变4个(表2)。结合心肌细胞的组织病理学特征,确定MYBPC3基因的T2155C(p.C719R)杂合突变为主要致病突变。图3为该突变的Sanger法测序验证。
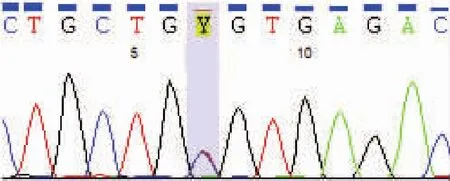
图3 MYBPC3的T2155C杂合突变的Sanger测序结果(Y代表C/T双峰)
3 讨论
本例猝死病例的心脏质量为550 g,室间隔厚度/左室壁厚度大于1.3,镜检可见心肌细胞肥大,多处心肌纤维化。结合死者在劳动中猝死的表现,按照HCM的病理学诊断标准[5],可从组织病理学水平确定其死因为HCM导致的心源性猝死。
在对死者基因组的全外显子组序列进行二代测序分析中,在外显子区域共检出181 418个SNV(包括SNP)。经过筛选后,最终筛选出4个可疑致病突变(表2)。结合心肌细胞的病理改变特征,最终确定MYBPC3基因的C719R的杂合突变为该病例的主要致病突变。
MYBPC3基因是HCM的最主要致病基因之一,约20%~30%的HCM是该基因突变所致[6]。MYBPC3基因(gi:Y10129)长约21 kb,mRNA长4 586 bp,包括35个外显子。在心肌细胞中编码1 274个氨基酸残基的蛋白cMYBPC,即心脏型肌球蛋白结合蛋白C,相对分子量在140 000左右。不同cMYBPC分子之间相互结合形成环状,垂直于肌丝的长轴分布,起到捆扎粗肌丝的作用[7]。cMYBPC分子由7个Ig模块和3个FnIII模块组成,排列顺序为C1-C5、C6、C7、C8、C9、C10(C6、7、9为FnIII模块)。C1和C2之间的一段105aa的结构域是cMYBPC分子负责与肌球蛋白myosin结合的结构域。一个分子cMYBPC通过其C5结构域与另一个分子cMYBPC的C8结构域结合,形成稳定结构。cMYBPC分子不仅参与正常肌小节和肌丝的组装,起到稳定肌小节结构的作用,而且通过磷酸化等调节横桥循环参与肌肉的收缩和舒张[8]。因此,cMYBPC分子突变势必对上述生理功能产生不同程度的影响,这也是MYBPC3基因突变导致HCM的生理基础。NIIMURA等[9]曾在一HCM家系中检测到MYBPC3基因22号外显子的1 bp的缺失突变,该突变发生的位置与本例HCM的C719R位置很近,均处于C5结构域,靠近富含脯氨酸的环结构。因此,推测C719R突变的结果(半胱氨酸转变为精氨酸)不仅涉及中性氨基酸转变为碱性氨基酸,也由小分子转变为较长的分子,这些改变会一定程度地影响两个MYBPC3分子的结合(C5结构域与C8结构域),进而影响肌小节结构稳定性,从而导致本例HCM的病变。具体的致病机制尚需要通过细胞和动物实验来证实。
MYBPC3基因突变形式多样,包括错义突变、插入或者缺失突变、剪接位点突变、无义突变等。截至2015年,已经报道了超过350种发生于MYBPC3基因的突变[10]。其中,约2/3突变涉及外显子序列移码突变或剪切位点突变,预测会产生长度异常的多肽链。其他突变则属于会产生氨基酸替代的各种单碱基非同义突变。据报道[11],MYBPC3基因突变导致的临床表型总体要好于其他几个肌节结构相关基因,如MYH7B、肌钙蛋白T(troponin T)和肌钙蛋白I(troponin I)等基因突变导致的临床表型。而且,携带插入或者缺失突变、剪接位点突变、无义突变等可以造成编码蛋白移码突变的HCM患者,较携带错义突变患者的预后要差,这也是MYBPC3基因突变不同于其他致病基因的重要特点。本研究病例的MYBPC3基因的C719R突变为错义突变,死者的猝死发生年龄较晚(48岁)及心脏质量(550 g)较大,均表明该突变早期的临床表型较温和,心肌经历了较长时间的功能代偿,其肥厚增大程度较高。因此,通过对其家族成员进行该基因突变的检测,可对该突变携带者进行早期诊断和干预,这对维护他们的健康生命以及优生优育具有重要意义。
本次致病突变筛选,还筛选出另外两个罕见的突变体(表2):RYR2基因的C3355T(p.R1119C)突变和TTN基因的C61057T(p.R20353C)突变。RYR2基因所编码的产物是内质网上一种钙离子释放通道。目前已发现超过100个RYR2基因突变与儿茶酚胺性多形性室性心动过速(catecholaminergic polymorphic ventricular tachycardia,CPVT)和心律失常性右室发育不良(arrhythmogenic right ventricular dysplasia,ARVD)等疾病相关,且已发现的突变主要集中于肽链的三个热点区:N端,中心段(FKBP12.6结合区)以及C端跨膜区[12,13]。本研究发现,C3355T(p.R1119C)不位于上述的突变热点区,因此,我们推测其对RY2分子的功能影响是非致命性的。同时,死者生前健康状况及本次病理学检验,均未发现死者有CPVT、ARVD等疾病的证据。因此,针对本例猝死病例,可排除该突变为主要的致命性突变,但不能排除这个突变可能会对猝死的发生起到一定的促进作用。
TTN基因包含364个外显子,编码肌联蛋白,是横纹肌细胞内肌小节结构中最大的分子。其编码产物N端连接肌小节的Z带,C端连接M带,起着调节肌节长度和弹性的生理功能。目前已经报道的TTN基因致病性突变有100多个[14],发生在心肌的突变常见于扩张型心肌病(dilated cardiomyopathy,DCM)病例,例如VAN SPAENDONCK-ZWARTS[15]、PUGH[16]等的研究。本研究的病例发现存在TTN基因的C61057T(p.R20353C)突变,属于新发现的突变(多态性)类型。从死者的心肌结构特征来看,心腔无明显扩张、心肌肥厚,缺乏DCM的典型征象。因此我们认为,TTN基因的C61057T(p.R20353C)突变对本例心肌病的发病原因贡献不大,该突变更应视为罕见多态性。
综合分析,本例猝死病例的死因为HCM导致的心源性猝死,其HCM的主要发病原因是MYBPC3基因的C719R的杂合突变。利用二代测序平台进行全外显子组测序,可快速筛选出怀疑有遗传基因突变机制的致病基因突变,为明确遗传性心肌病猝死病例的分子病变机制提供快捷准确的检测技术。
[1]GERSH B J,MARON B J,BONOW R O,et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy:executive summary:a report of the american college of cardiology foundation/american heart association task force on practice guidelines[J].Circulation,2011,124(24):2761-2796.
[2]ELLIOTT P,MCKENNA W J.Hypertrophic cardiomyopathy[J].Lancet,2004,363(9424):1881-1891.
[3]MARON B J,MARON M S,SEMSARIAN C.Genetics of hypertrophic cardiomyopathy after 20 years:clinical perspectives[J].J Am Coll Cardiol,2012,60(8):705-715.
[4]王纯,王辉,许心舒,等.外显子组测序对1例青壮年猝死的分子遗传学分析[J].法医学杂志,2015,31(6):436-440,444.
[5]李玉林.病理学[M].8版.北京:人民卫生出版社,2013.
[6]MARIAN A J.Hypertrophic cardiomyopathy:from genetics to treatment[J].Eur J Clin Invest,2010,40(4):360-369.
[7]WINEGRAD S.Cardiac myosin binding protein C[J]. Circ Res,1999,84(10):1117-1126.
[8]FLASHMAN E,REDWOOD C,MOOLMANSMOOK J,et al.Cardiac myosin binding protein C:its role in physiology and disease[J].Circ Res,2004,94(10):1279-1289.
[9]NIIMURA H,BACHINSKI L L,SANGWATANAROJ S,et al.Mutations in the gene for cardiac myosinbinding protein C and late-onset familial hypertrophic cardiomyopathy[J].N Engl J Med,1998,338(18):1248-1257.
[10]CARRIER L,MEARINI G,STATHOPOULOU K,et al.Cardiac myosin-binding protein C(MYBPC3)in cardiac pathophysiology[J].Gene,2015,573(2):188-197.
[11]RICHARD P,CHARRON P,CARRIER L,et al. Hypertrophic cardiomyopathy:distribution of disease genes,spectrum of mutations,and implications for a molecular diagnosis strategy[J].Circulation,2003,107(17):2227-2232.
[12]BLAYNEY L M,LAI F A.Ryanodine receptormediated arrhythmias and sudden cardiac death[J]. Pharmacol Ther,2009,123(2):151-177.
[13]MEDEIROS-DOMINGO A,BHUIYAN Z A,TESTER D J,et al.Comprehensive open reading frame mutational analysis of the RYR2-encoded ryanodine receptor/calcium channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative,exerciseinduced long QT syndrome[J].J Am Coll Cardiol,2009,54(22):2065-2074.
[14]CHAUVEAU C,ROWELL J,FERREIRO A.A rising titan:ttn review and mutation update[J].Hum Mutat,2014,35(9):1046-1059.
[15]VAN SPAENDONCK-ZWARTS K Y,POSAFALVI A,VAN DEN BERG M P,et al.Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy[J].Eur Heart J,2014,35(32):2165-2173.
[16]PUGH T J,KELLY M A,GOWRISANKAR S,et al.The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing[J].Genet Med,2014,16(8):601-608.
Gene Analysis for the Sudden Death of Hypertrophic Cardiomyopathy by Whole Exome Sequencing
XU Chuan-chao1,2,BAI Yun-zhi3,XU Xin-shu4,LÜGuo-li4,LAI Xiao-ping1,2,CHEN Rui1,2,LIN Hanguang1,2,KUANG Wen-jian1,2
(1.Guangdong Medical University,Dongguan 523808,China;2.Center of Forensic Science,Guangdong Medical University,Dongguan 523808,China;3.Panyu Branch of Guangzhou Public Security Bureau,Guangzhou 511400,China;4.Guangzhou Institute of Forensic Science and Technology,Guangzhou 510030,China)
Objective To analyze the related pathogenicity gene mutations in a sudden death of hypertrophic cardiomyopathy(HCM)on whole exome level.Methods Whole exome sequencing(WES)was been performed on a sudden death case sample with pathological features of HCM by Illumina®Hiseq 2500 platform.Using hg19 as the reference sequences,the sequencing data were analyzed.Suspicious single nucleotide variants(SNV)were screened,and the conservatism and function were analyzed by the software such as PhyloP,PolyPhen-2,SIFT,etc.Results After screening,a heterozygous mutation C719R was finally identified in the gene MYBPC3 of this case.Conclusion The molecular anatomy on whole exome level by second generation sequencing technology can help to define the molecular mechanism of HCM and provide a new mothed and thought for analysis of death cause.
forensic pathology;cardiomyopathy,hypertrophic;death,sudden,cardiac;exome;mutation
DF795.1
A
10.3969/j.issn.1004-5619.2017.04.001
1004-5619(2017)04-0339-05
2016-03-03)
(本文编辑:邹冬华)
2013年广东省自然基金资助项目(S2013040011977);2015年度广东省自然科学基金资助项目(2015A030310456)
许传超(1972—),男,讲师,主要从事分子遗传学研究;E-mail:xcchao5855@sina.com
赖小平,男,副教授,主要从事法医病理学研究;E-mail:g_xplai@163.com
