基于离子液体基质的大豆中寡糖成分基质辅助激光解吸电离—质谱成像分析
2017-08-14裴兴丽黄煜宇龚灿许旭
裴兴丽+黄煜宇+龚灿+许旭


摘 要 将离子液体2,5-二羟基苯甲酸丁胺用于改进基质辅助激光解吸电离质谱分析寡糖的定量重复性,并进一步用于大豆和豆叶中寡糖的质谱成像研究。实验中设定正电荷检测模式、激光能量为70%,采用将离子液体2,5-二羟基苯甲酸丁胺甲醇溶液(20%,V/V)直接覆盖样品的简单加基质方法,分析寡糖样品及大豆和豆叶寡糖分布的质谱成像。结果表明,离子液体2,5-二羟基苯甲酸丁胺作为基质,用基质辅助激光解吸电离化质谱分析蔗糖、棉子糖、水苏糖3种寡糖样品,点内重复性RSD<3%,点间重复性RSD<4%,在0.062~1.00 mg/mL 范围内测定的线性相关系数R2≥ 0.996,显示出较好的定量分析潜力。将此基质用于基质辅助激光解吸电离质谱成像分析大豆切片和豆叶表面的二糖、三糖和四糖,得到空间分辨率为150 μm的3种寡糖质谱成像图。3种寡糖在大豆中大致均衡分布,在豆叶的分布均以叶尖为多,并且根据标准曲线和图中的信号强度可以估计其含量。
关键词 质谱成像; 离子液体; 基质辅助激光解吸; 寡糖
1 引 言
基质辅助激光解吸电离质谱(Matrix-assisted laser desorption ionization-mass spectrometry, MALDI-MS)成像技术是分析组织或不同组织状态之间组分变化非常有价值的工具,近年得到快速发展[1,2],已广泛应用于病理学[3]、药物代谢[4]、动植物生理[5]等诸多生命科学领域。该技术在分子水平上对生物组织切片进行多组分“直接”分析,经过图像重建软件获得组织中目标成分的定位、定性和定量的信息,并提供可视化的组织分布信息[6,7]。
由于質谱成像中包含样品的含量信息,这也需要解决MALDI-MS用于定量分析的问题,目前已有不少实验室用不同方法对各种样品进行定量分析[8]。这些工作内容大致可以分为四方面: 一是优化实验参数,通过控制样品制备和预处理过程降低测量差异[9,10]; 二是从分析方法入手,采用同位素稀释法[11]、内标法定量[12,13]、定量为与已知内源性组分的比值[14]等; 三是改善基质覆盖均匀性,如喷雾-液滴法[15],化学喷墨打印法[16]等; 四是选择不同的基质,张珍英等[17]采用新型香豆素类化合物分别与2,5-二羟基苯甲酸(2,5-Dihydroxybenzoic acid, DHB)混合组成两种二元基质,显著改善了基质和葡聚糖样品的共结晶状况,使样品分布更均匀。Jiao等[18]采用一种新型肼基烟酸做基质分析低聚糖,发现比传统的基质DHB重复性好。MALDI-MS成像和定量分析的关键与选择合适的基质密切相关。传统的单一固体基质容易形成热点(Hot spot)导致结晶不均匀、信号重复性差等问题,其样品点内(In-spot)、点与点之间(Spot-to-spot)等的重复性仍然存在不少问题[19]。
离子液体(Ionic liquid, IL)是在室温下呈液态, 是由离子构成的有机盐[20]。Armstrong等[21]首次将离子液体引入MALDI-MS作为基质,显示出灵敏度高、真空稳定性好、能与样品均匀混合、重复性好等优点。进一步使用离子液体的MALDI-MS研究[22~24]对多种样品获得了良好的重复性、线性和灵敏度。使用α-氰基-4-羟基肉桂酸(α-Cyano-4-hydroxycinnamic acid, CHCA)或DHB与等摩尔有机碱(如三丁胺、吡啶和1-甲基咪唑等)得到的离子液体作为MALDI基质,具有可忽略的蒸气压,特别是其与样品形成均匀的混合物表面,减少了点与点之间的信号差异, MALDI-MS的定量重复性明显改善[25~27]。如Yoon等[28]采用CHCA与1-甲基咪唑得到的离子液体基质对聚六甲基胍样品进行定量分析。将离子液体用于MALDI-MS成像也显示出较好的成像效果。Chan等[29]将CHCA与1-甲基咪唑的离子液体基质用于MALDI-MS成像分析小鼠脑中神经节苷脂的分布。Shrivas等[30]将CHCA丁胺和DHB丁胺离子液体基质用于小鼠肝组织和脑部切片中磷脂的MALDI-MS成像分析。 Meriaux等[31]采用DHB和苯胺、DHB和3-乙酰合成的离子液作为基质,用于MALDI-MS成像分析大鼠脑部和人卵巢癌的脂质。
寡糖具有特殊的生理功能,不但对促进植物生长、增强抗逆性等方面有重要作用,在疾病防治、植物生长与抗病害、畜牧养殖等方面的应用也备受关注[32]。
本研究以离子液体2,5-二羟基苯甲酸丁胺(2,5-Dihydroxybenzoate acid butylamine, DHB-BuN)为基质,使用傅里叶变换离子回旋共振质谱,MALDI-MS分析寡糖样品,在获得MALDI-MS较好定量重复性和灵敏度的基础上,将此基质用于MALDI-MS成像分析大豆切片和大豆叶片表面的二糖、三糖和四糖,为寡糖的MALDI-MS成像分析提供了实验参考。
2 实验部分
2.1 仪器与试剂
SolariX 7.0 型傅立叶变换离子回旋共振质谱(FT-ICR-MS,瑞士 Bruker公司),配备Nd:YAG二级管泵浦固体激光器(波长为355 nm)和HyStar Version 3.4(Build 8)成像分析软件; BS224S 型电子天平(北京赛多利斯仪器系统有限公司); PS-20超声波清洗仪(东莞洁康公司); XW-80A旋涡混合仪(海门市其林贝尔仪器制造有限公司); ZX98-1旋转蒸发器(上海豫康科教仪器设备有限公司); Finesse325型旋转石蜡切片机(赛默飞世尔科技(上海)有限公司)。
甲醇、乙腈、吡啶、苯胺、N,N-二甲基苯胺、正丁胺、D-(+)-蔗糖(分析纯,上海泰坦科技股份有限公司)。L-脯氨酸(生化试剂)、D,L-焦谷氨酸(纯度98%)、L-精氨酸盐酸盐(生化试剂)、D,L-酪氨酸(纯度98.5%)均购自国药集团化学试剂有限公司。2,5-二羟基苯甲酸(DHB,纯度为99%)、水苏糖(纯度为98%)、D-(+)-葡萄糖(纯度99%)均购自北京百灵威科技技术有限公司。β-环糊精(纯度≥98%,上海源叶生物科技有限公司)。棉子糖(纯度98%,上海同田生物技术股份有限公司)。谷胱甘肽(还原型,纯度99%,如吉生物科技有限公司)。血管紧张素Ⅱ(纯度90%。阿拉丁试剂)。L-苯丙氨酸(纯度98%,Adamas试剂有限公司)。蒸馏水和葵花籽油购自本地超市。大豆(当年收获,产地: 江苏大丰),豆叶采自本地当季豆田。
2.2 实验方法
2.2.1 基质配制与样品制备 (1)离子液体基质的合成 参考文献[33]方法,取2,5-二羟基苯甲酸置于圆底烧瓶,加入甲醇溶解,配成0.2 mol/L 的溶液。加入稍过量的正丁胺或者其它有机胺,混合1 min, 旋蒸(40℃)除去多余的丁胺和甲醇,直到液体体积不再减少,得到纯的离子液体2,5-二羟基苯甲酸丁胺(DHB-BuN)。直接加入4倍体积的甲醇并涡旋10 min,配成20% (V/V) 的溶液,待用。合成的基质在2天内使用,或者充入氮气(避免氧化)密封保存。(2)固体基质的制备 固体基质2,5-二羟基苯甲酸(DHB)用乙腈-水(7∶3, V/V)配成浓度为10 mg/mL的溶液。(3)样品准备 取适量蔗糖(二糖)、棉子糖(三糖)、水苏糖(四糖)、β-环糊精、脯氨酸、焦谷氨酸、L-精氨酸、L-苯丙氨酸、D,L-酪氨酸、血管紧张素Ⅱ、谷胱甘肽、葵花籽油,分别溶于水-乙腈(1∶1, V/V)溶液,配制成0.5 mg/mL的溶液。(4)材料的处理 大豆: 用石蜡切片机切成厚度为25 μm的薄片,切片位置离胚芽约0.3 mm,垂直切下,然后取切下的一半。豆叶: 摘下的豆叶,用剪刀剪下尖端部分(得到面积约2 cm2的三角形)待用。分析时用双面胶分别贴在不锈钢靶板上。
2.2.2 仪器条件 (1)MALDI-MS条件 点靶后采用正离子模式检测,使用离子液体时分析样品的采集与成像参数如表1所示。固体基质DHB的采集参数除激光能量为45%外,其它与使用离子液体基质分析的采集参数相同。(2)MALDI-MS成像实验 质谱成像的过程如图1所示。首先将样品薄片用双面胶直接贴在不锈钢靶板上(或者贴在成像玻片上,并装入成像靶板),然后采用干滴法,用移液枪取制备好的的离子液体基质滴在样品薄片上,待溶剂挥发后,放入质谱仪。在Compass solariXcontrol质谱操作软件中设置采集参数见表1。成像结果由Hystar Version 3.4(Build 8))软件处理得到。
3 结果与讨论
3.1 MALDI-MS实验条件的影响
选用葡萄糖(单糖)、蔗糖(二糖)、棉子糖(三糖)、水苏糖(四糖)、β-环糊精(七糖)作为寡糖样品。样品浓度均为0.5 mg/mL。考察了不同基质类型、基质溶液浓度、基质溶液与样品溶液用量比、激光能量、点样方法等对测试的影响。
用2,5-二羟基苯甲酸(DHB)分别与吡啶、苯胺、N,N-二甲基苯胺、正丁胺合成了4种离子液体基质。用于3种寡糖样品测定时均有响应,其中DHB与正丁胺形成的离子液体DHB-BuN作为基质对3种寡糖的响应均较好。因此,选择DHB-BuN作为进一步研究的基质。
考察DHB-BuN甲醇溶液浓度分别在14%~50%(V/V)时的MALDI-MS点样效果,在DHB-BuN浓度低于20%(V/V)时, 溶液在靶板上能均匀铺展,不易聚集,得到均匀且薄的基质点。但降低浓度,信噪比也会下降。故DHB-BuN甲醇溶液浓度选择20%(V/V)。
以蔗糖为样品,考察在基质溶液与样品溶液的用量(DHB-BuN∶蔗糖)分别为2∶1、1∶1、1∶2(V/V)时的测试结果,当基质与样品溶液的体积比为1∶1时,得到的质谱峰信噪比较好。
当激光能量低于55%,样品不出峰; 考虑到能量大于80%可能影响激光寿命,所以激光能量选择为70%。
比较了3种点样方法,包括(A)先用基质DHB-BuN甲醇溶液 0.5 μL点靶,然后点样0.5 μL蔗糖溶液(浓度0.5 mg/mL); (B)先取样品蔗糖溶液(0.5 mg/mL)0.5 μL 点样,然后用基质DHB-BuN溶液 0.5 μL 點样; (C)基质DHB-BuN溶液和样品蔗糖溶液(0.5 mg/mL)等体积混合后超声1 min,再涡旋2 min充分混匀后,取1.0 μL点靶。用MALDI-MS成像的方法(实验参数见表1)评价组分在靶板上分布的均匀性。结果表明,基质-样品混合后点样重复性较好,具有最好的均匀性。
因此,实验中将20%(V/V)的DHB-BuN甲醇溶液和样品溶液等体积混合后超声1 min,再涡旋2 min充分混匀后,取1.0 μL点靶,按照2.2.2节MALDI-MS方法测定。5种寡糖样品测试结果与理论值的比较见表2,除葡萄糖以[M+Na]+的形式出峰外,其它成分都以[M+Na]+、[M+K]+的形式出峰。
3.2 重复性
选择混合点样方法(C),将上述配制的浓度均为0.5 mg/mL的蔗糖、棉籽糖、水苏糖分别与基质混合,在以上MALDI-MS条件下,分别用离子液体基质DHB-BuN和固体基质DHB测试样品,比较两者的重复性。在比较点内(In-spot)重复性时,按照九宫格的形式,将1个点分成大致9个区域,然后手动将激光束聚焦在每个区域的位置上,采集质谱数据。选择[M+Na]+峰为重复性数据的目标峰,每个区域的数据是重复采集区内不同位置的5次样品信号强度的平均值(采用DHB-BuN时,RSD<6%; 采用DHB时,RSD<15%)。点间(Spot-to-spot)的重复性是测定6个不同样品点的结果,在每个样品点中间区域的不同位置采集5次数据的平均值。由图2可见,采用DHB-BuN基质测定的点内峰强度RSD<3%,点间RSD<4%,可较好地进行定量分析和成像研究。
采用MALDI-MS成像方法测定靶上样品在不同区域的信号强度分布,靶点的光学图像和MS成像如图3所示。从DHB基质的光学图像(A)可以看到产生了结晶,而DHB-BuN做基质时的光学图像(D)中可以看到DHB-BuN仍然以液体状态存在。在质谱仪高倍放大镜头下的分布见图3B和3E。MALDI-MS成像的方法测定靶上样品在不同区域的信号强度分布见图3C和3F。采用DHB-BuN基质时的信号强度分布明显比DHB为基质时样品信号强度分布更均匀,这与文献[34]和图2的结果一致。
3.3 质谱信号强度与样品浓度间的(线性)关系
分别将6种不同浓度的(0.062~1.000 mg/mL)蔗糖、棉籽糖、水苏糖样品与基质混合超声超1 min, 再用旋涡混合2 min,使基质和分析物充分混匀。用移液枪分别取1 μL各浓度混合溶液,分别点在靶板上,MALDI-MS测定质谱峰强度。测定样品浓度与[M+Na]+峰强度的线性关系(图4),其中每个浓度的峰强度同样是重复采集5次数据求平均值得到(5次质谱峰强度数据RSD<6%)。在本实验测定的浓度范围内各寡糖样品的质谱峰信号强度与样品浓度, 线性关系良好,相关系数R2≥ 0.996。
3.4 灵敏度
用离子液体基质DHB-BuN和固体基质DHB(10 mg/mL)分别测试浓度为0.5 mg/mL的蔗糖、棉子糖、水苏糖标准样品的MALDI-MS谱图(图5)。不同基质测得各样品信号强度和信噪比见表3,DHB-BuN相对固体基质DHB的信号强度、基线噪音、信噪比均有不同程度的提高,蔗糖、棉子糖和水苏糖的信噪比分别提高了约2倍、8.4倍和5.5倍。按照3倍信噪比计算,各寡糖样品的检出限为7~25 μg/mL。 表明离子液液体基质DHB-BuN具有较高的灵敏度。
在测试标准样品和实际样品时,标准样品主要以[M+Na]+出峰,而实际测大豆样品时,主要以 [M+K]+出峰。因此考察了加入Na+和K+对信号强度的影响。首先在标准样品(1.0 mg/mL)中加入不同浓度的盐(NaCl或者KBr),测定结果见图6,
当加入KBr的浓度低于8 mmol/L时, [M+K]+的信号响应增强; 但KBr的浓度超过8 mmol/L后,样品的信号强度降低。加入NaCl溶液浓度在低于7 mmol/L时,有助于增强 [M+Na]+的信号; NaCl浓度超过7 mmol/L时,信号强度也降低。因此,加入适量的Na+和K+会增强样品峰强度,但浓度高则有抑制作用。大豆样品中的[M+K]+峰则可能来自于样品中的K+。
以DHB-BuN为基质,MALDI-MS测试了脯氨酸、焦谷氨酸、L-精氨酸、L-苯丙氨酸、D,L-酪氨酸、血管紧张素Ⅱ、谷胱甘肽和葵花籽油样品。结果表明,只有苯丙氨酸、谷胱甘肽和血管紧张素Ⅱ有质谱峰出现,其它样品均不出峰。进一步考察苯丙氨酸、谷胱甘肽和血管紧张素Ⅱ对寡糖测定的影响,将蔗糖和血管紧张素Ⅱ配成等摩尔浓度的溶液,然后以两种不同的体积比混合(1∶1~1∶10),MALDI-MS测定结果见图7,当蔗糖-血管紧张素Ⅱ的体积比为1∶10时,蔗糖仍有很强的峰,而同样摩尔浓度的血管紧张素Ⅱ质谱峰的强度仅约为蔗糖质谱峰强度的1/4,如图7B所示。说明DHB-BuN基质能选择性地使低聚糖离子化,而此时对血管紧张素Ⅱ类的离子化能力不强。同样测试苯丙氨酸,也有类似现象出现。但采用蔗糖+谷胱甘肽测试时,两者的峰强度相近,寡糖的峰强度与单独测试时基本相同。因此,离子液体作为基质时,对不同的氨基酸或者多肽样品的MALDI-MS响应存在明显差异,在本实验条件下,不会影响对寡糖的质谱分析。
与常规的基质(如DHB)相比,离子液体基质也需从紫外吸收基团获取激光的能量,实验中还需使用较高的激光能量,这可能与样品离子化时需要克服液体的表面张力有关。对于多肽等样品,本方法的检测灵敏度比固体基质有不同程度的提高(这也与其能量偏高有关)。文献中也报道测定寡核苷酸[35]、磷脂[36]样品时有灵敏度提高的现象。
3.6 大豆中的寡糖的成像
将大豆用石蜡切片机切成厚度为25 μm的薄片。按照2.2.2节的方法,取30 μL DHB-BuN,分3次(每次10 μL)尽可能均匀地滴在大豆上,得到的大豆切片的MAILDI-MS结果如图8所示。本实验的质谱条件无法区分同分异构体,大豆中主要的寡糖为二糖(蔗糖)、三糖(棉子糖)和四糖(水苏糖),与文献[37]报道一致。且这些二糖、三糖、四糖主要是[M+K]+峰,[M+Na]+峰很低。
MALDI-MS成像分辨率介于二次離子质谱(Secondary ion mass spectrometry, SIMS)成像与常压敞开式离子化质谱成像技术之间[38],本实验的仪器可以使用25 μm的空间分辨率,但耗时较长,通常仅在局部样品分析时使用。本实验中设置150 μm空间分辨率,由Hystar软件测试得到大豆切片(8.0 mm×3.4 mm)的MALDI-MS成像图见图9,各种寡糖在该切片中大致均匀分布,但不同寡糖的分布不完全一样。根据前面的线性关系和质谱数据,可以大致估计不同寡糖的最高浓度(粉色区域)分别约为130 μg/mL(二糖)、25 μg/mL(三糖)和100 μg/mL(四糖)。
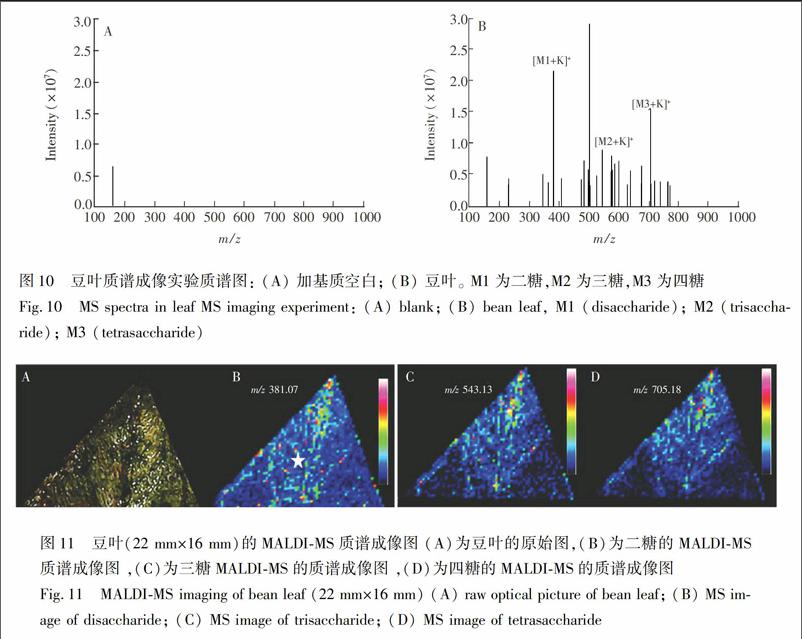
对豆叶的质谱成像测定时同样采用2.2.2节的方法,取30 μL DHB-BuN溶液,分3次(每次10 μL)尽可能均匀地滴在豆叶样品(底宽22 mm,高16 mm)上,MALDI-MS测豆叶样品表面的质谱图如图10所示, 豆叶中的寡糖的质谱峰都是[M+K]+峰。MALDI质谱成像如图11所示。根据3.3节的线性关系,可大致推算最高浓度(粉色区域)分别为300 μg/mL(二糖)、76 μg/mL(三糖)和40 μg/mL(四糖)。其中蓝色区域的含量基本在检出限附近。各寡糖在豆叶的分布均以叶尖为多。
从表1可知, DHB-BuN为基质可测定出单糖(葡萄糖)和七糖(β-环糊精),但在豆叶的质谱中并没有发现除二糖、三糖、四糖以外的其它单糖和寡糖的质谱峰。本方法测定单糖(葡萄糖)的检出限为0.1 mg/mL, β-环糊精的检出限为16 μg/mL。样品中没有测出单糖和其它寡糖,说明大豆和豆叶表面这些成分的含量低于检出限。
4 结 论
离子液体基质DHB-BuN用于MALDI-MS分析寡糖样品,操作简便,重复性和灵敏度优于固体基质,可用于寡糖的MALDI-MS定量和成像分析。本研究将此离子液基质用于MALDI-MS成像,获得了大豆和豆叶中寡糖分布的质谱成像图。实验中的大豆切片内主要为二糖、三糖和四糖,且分布较为均匀; 豆叶中也仅测出这3种寡糖,且在叶尖部位较多。本方法无需复杂的样品前处理过程,能够直观反映寡糖的分布和浓度等原位信息,质谱检测背景干扰低。离子液体基质DHB-BuN对寡糖类物质的MALDI-MS分析在定量和MALDI-MS成像方面具有较好的应用潜力。
References
1 Chughtai K, Heeren R M A. Chem. Rev., 2010, 110(5): 3237-3277
2 Holzlechner M, Strasser K, Zareva E, Steinhauser L, Birnleitner H, Beer A, Bergmann M, Oehler R, Marchetti-Deschmann M. J. Proteome. Res., 2017, 16(1): 65-76
3 Ly A, Schne C, Becker M, Rattke J, Meding S, Aichler M, Suckau D, Walch A, Hauck S M, Ueffing M. Histochem. Cell Biol., 2015, 143(5): 453-462
4 Niehoff A C, Kettling H, Pirkl A, Chiang Y N, Dreisewerd K, Yew J Y. Anal. Chem., 2014, 86(22): 11086-11092
5 Beck S, Stengel J. Phytochemistry, 2016, 130: 201-206
6 Pirman D A, Reich R F, Kiss A, Heeren R M A, Yost R A. Anal. Chem., 2013, 85(2): 1081-1089
7 Lagarrigue M, Lavigne R, Tabet E, Genet V, Thomé J P, Rondel K, Guével B, Multigner L, Samson M, Pineau C. Anal. Chem., 2014, 86(12): 5775-5783
8 LI Xin-Xin, CHEN Lin-Fei, OUYANG Yong-Zhong, FENG Fang, CHEN Huan-Wen. Chinese J. Anal.Chem., 2016, 44(1): 25-31
李欣昕, 陈林飞, 欧阳永中, 冯 芳, 陈焕文. 分析化学, 2016, 44(1): 25-31
9 Heeren R M A, Kǜkrer-Kaletas B, Taban I M, MacAleese L, McDonnell L A. Appl. Surf. Sci., 2008, 255(4): 1289-1297
10 Goodwin R J A, Pennington S R, Pitt A R. Proteomics, 2008, 8: 3785-3800
11 Kuklenyik Z, Boyer A E, Lins R, Quinn C P, Candela M G, Woolfitt A, Pirkle J L, Barr J R. Anal. Chem., 2011, 83(5): 1760-1765
12 Prentice. B M, Chμmbley C W, Caprioli R M. J. Am. Soc. Mass Spectrom., 2017, 28: 136-144
13 David A P, Richard A Y. Anal. Chem., 2011, 83(22): 8575-8581
14 Hattan S J, Parker K C, Vestal M L, Yang J Y, Herold D A, Duncan M W. J. Am. Soc. Mass Spectrom., 2016, 27: 532-541
15 Sugiura Y, Shimma S, Setou M. Anal. Chem., 2006, 78(24): 8227-8235
16 Baluya D L, Garrett T J, Yost R A. Anal. Chem., 2007, 79(17): 6862-6867
17 ZHANG Zhen-Ying, DENG Hui-Min, DENG Qin-Ying. Chinese J. Anal. Chem., 2008, 36(10): 1419-1422
張珍英, 邓慧敏, 邓芹英. 分析化学, 2008, 36(10): 1419-1422
18 Jiao J, Zhang Y, Yang P, Lu H. Analyst, 2015, 140(1): 156-161
19 Duncan M W,Roder H, Hunsucker S W. Genomics Proteomics, 2008, 7(5): 355-370
20 Ho T D, Zhang C, Hantao L W, Anderson J L. Anal. Chem., 2014, 86(1): 262-285
21 Armstrong D W, Zhang L K, He L, Gross M L. Anal. Chem., 2001, 73(15): 3679-3686
22 Shrivas K, Tapadia K. J. Chromatogr. B, 2015, 1001: 124-130
23 KUANG Min, ZHANG Ying, YANG Peng-Yuan, LU Hao-Jie. Acta Chim. Sinica, 2013, 71: 1007-1010
匡 敏, 张 莹, 杨芃原, 陆豪杰.化学学报, 2013, 71: 1007-1010
24 Zabet-Moghaddam M, Heinzle E, Tholey A. Rapid Commun. Mass Spectrom., 2004, 18(2): 141-148
25 Mank M, Stahl B, Boehm G. Anal. Chem., 2004, 76(10): 2938-2950
26 Li Y L, Gross M L. J. Am. Soc. Mass Spectrom., 2004, 15(12): 1833-1837
27 Fukuyama Y, Nakaya S, Yamazaki Y, Tanaka K. Anal. Chem., 2008, 80(6): 2171-2179
28 Yoon D, Lee D, Lee J H, Cha S, Oh H B. Rapid Commun. Mass Spectrom, 2015, 29: 213-219
29 Chan K,Lanthier P, Liu X, Sandhu J K, Stanimirovic D, Li J. Anal. Chim. Acta, 2009, 639: 57-61
30 Shrivas K, Hayasaka T, Goto-Inoue N, Sugiura Y, Zaima Y, Setou M. Anal. Chem., 2010, 82 (21): 8800-8806
31 Meriaux C, Franck J, Wisztorski M, Salzet M, Fournier I. J. Proteomics, 2010, 73(6): 1204-1218
32 ZHANG Yun-Hong, WU Li-Shu, GENG Ming-Jian, XU Fang-Sen, ZHANG Shan-Xue. Plant Physiology Communications , 2009, 45(12): 1239-1245
张运红, 吴礼树, 耿明建, 徐芳森, 张善学. 植物生理学通讯, 2009, 45(12): 1239-1245
33 Mank M, Stahl B, Boehm G. Anal. Chem., 2004 , 76(10): 2938-2950
34 McCombie G, Knochenmuss R. Anal. Chem., 2004, 76: 4990-4997
35 Carda-Broch S, Berthod A, Armstrong D W. Rapid Commun. Mass Spectrom., 2003, 17(6): 553-560
36 Li Y L, Gross M L, Hsu F F. J. Am. Soc. Mass Spectrom., 2005, 16(5): 679-682
37 HUANG Xian-Xiao, GU Ke-Ren, ZHAO Yi-Fan. Cereal and Food Industry, 2006, 13(3): 27-31
黃贤校, 谷克仁, 赵一凡. 粮食与食品工业, 2006, 13(3): 27-31
38 LUO Zhi-Gang, HE Jiu-Ming, LIU Yue-Ying, LI Tie-Gang, HE Jing-Jing, ZHANG Si-Chun, ZHANG Xin-Rong, ABLIZ Zeper. Scientia Sinica Chimica, 2014, 44(5): 795-800
罗志刚, 贺玖明, 刘月英, 李铁钢, 何菁菁, 张四纯, 张新荣, 再帕尔·阿不力孜. 中国科学: 化学, 2014, 44(5): 795-800
