一株金黄色葡萄球菌小菌落突变株全基因组测序分析
2017-05-18王奇惠时永强张赛飞朱立力曲伟杰
王奇惠,时永强,祝 宇,张赛飞,朱立力,曲伟杰
(云南农业大学动物科学技术学院,云南昆明 650201)
一株金黄色葡萄球菌小菌落突变株全基因组测序分析
王奇惠,时永强,祝 宇,张赛飞,朱立力,曲伟杰*
(云南农业大学动物科学技术学院,云南昆明 650201)
对金黄色葡萄球菌小菌落突变株(small colony mutant strainsStaphylococcusaureus,SASCVs)和CP000253.1基因组差异进行了研究,为后续研究金葡菌分泌抗药物相关物质能力提供数据支持。对一株金葡菌小菌落突变株进行全基因组测序,并与CP000253.1参考序列进行序列比较。结果显示,金葡菌SCVs的文库插入片段为350 bp,测序读长为150 bp,SCVs的原始数据量为551 Mb,SCVs的碱基含量分布在处理前后有所不同,通过重测序,存在单核苷酸多态性(SNP)的数量较大。金葡菌SCVs与CP000253.1参考序列之间存在明显的序列差异,该数据为研究金葡菌感染奶牛乳房炎提供了一定的序列依据。
金葡菌小菌落突变株;基因序列差异;全基因组测序;单核苷酸多态性
奶牛乳房炎是奶牛最常见的疾病之一[1-2]。由金黄色葡萄球菌(Staphylococcusaureus)所引起的奶牛慢性乳房炎一直制约着我国奶牛养殖业的发展。细菌小菌落突变株(small colony variants,SCVs)于1910年首次被报道,当时分离的一株SCVs被认为是一种异常伤寒菌,后来被证实这株SCVs属于肠伤寒沙门菌。在临床上,SCVs 比相应的野生株更能持续存在于哺乳动物的细胞中,并且对抗生素不敏感,当宿主细胞形成应急保护性的环境时,SCVs会引起隐性或者复发性感染。在许多细菌种属中都发现有SCVs,但葡萄球菌SCVs研究的相对较少,尤其是金黄色葡萄球菌(简称金葡菌)。金葡菌 SCVs 的独特表型与持续复发性感染之间的联系在20世纪90年代中期才被认知[3-7]。近几年来,金葡菌SCVs可致人和动物严重感染。国内外相继报道了一类具有特殊表型的金葡菌小菌落突变株[4](small colony mutant strainsStaphylococcusaureus,SASCVs),并证明了它们与金葡菌感染所致奶牛慢性乳房炎的关系非常密切,这无疑为研究该病的详细致病机制提供了新的思路[5-6]。
全基因组测序是对已经公布了其基因组序列的物种进行其他个体的基因组测序,并在此基础上针对个体或群体进行差异性等分析[7]。全基因组测序的个体,通过把其测序的短序列比对到已知的基因组上,检测出全基因组上大量的单核苷酸多态性位点(SNP,插入缺失位点(In Del、结构变异位点(SV)位点等[8-9]。基于变异位点数据,通过生物信息手段,分析不同个体或群体基因组间的结构差异,同时完成注释[11-13]。
为了研究金葡菌SCVs的基因组序列情况,应用第2代测序方法[10]对1株金葡菌SCVs的基因组序列进行了全基因组测序,并比较其与CP000253.1参考序列差异,旨在发现金葡菌SCVs菌株特有的序列特征,为研究不同来源金葡菌之间功能的差异提供基础[14]。
1 材料与方法
1.1 材料
金葡菌SCVs分离自无菌采集的患病奶牛的奶样中,由本实验室保存。营养琼脂(10 g/L的胰蛋白酶,5 g/L的酵母粉,5 g/L的NaCl,15 g/L的琼脂)培养基37℃有氧培养18 h~24 h。
1.2 方法
1.2.1 基因组DNA提取及测序 基因组DNA用Ge-nomic-Tip500/G试剂盒(Qiagen Inc.,Valencia,CA,USA)试剂盒提取,10 mmol/L Tris-盐酸(pH8.5)重悬保存备用,经超声波破碎仪(Diageno-de SA,Liège,Belgium)处理(工作15 s,间隔90 s,重复3次),得到 DNA片段。使用Tru Seq DNA Sample Preparation试剂盒(Illumina Inc.,San Diego,CA,USA)构建基因组文库。
1.2.2 序列的比对和SNP分析 首先对原始数据进行过滤,提取高质量的数据(clean data)与参考序列进行比对,其次根据比对情况检测样品的SNP,并对SNP结果注释。
1.2.3 信息分析 数据处理,此步骤过滤测序质量值低的reads,保留高质量reads,过滤后的数据称为clean data;然后与参考基因组进行比对,通过序列比对的方法,将得到的clean data与参考基因组进行序列比对分析;再进行SNP分析,根据比对情况检测金葡菌SCVs的SNP,并对SNP结果注释。
2 结果
2.1 金葡菌SCVs菌株的全基因组测序结果
本研究利用第2代测序技术,使用Hiseq PE150测序平台对样品进行测序,各样品的数据经过数据质控,过滤低质量的序列片段。详细的质控测序统计信息结果如下:SCVs的文库插入片段(insert size)大小是350 bp(base pair,碱基对),测序长度(reads length)是150 bp:150 bp(冒号前后分别为read1和read2的长度)。SCVs的原始数据Raw data的数据量为551 Mb(Mega base pair,百万碱基对),其中被过滤掉的reads占raw data总reads的2.05%,clean data的数据量为504 Mb,其中有效数据的GC含量为34.55%,有效数据的Q20值为97.35%,有效数据的Q30值为93.1%,有效数据的碱基含量分布情况见图1。从SCVs的碱基含量分布图中可以看出,处理前碱基含量分布和处理后碱基含量分布并没有太大的差别。将杂质处理掉后,图中没有明显的变化,只有稍微些许变化。
应用SAMtools软件,以CP000253.1作为参考基因组,依照SNP和基因之间的位置关系及相互作用,对SNP功能进行注释。通过对SCVs的SNP注释结果统计(图2),通过与CP000253.1参考序列进行比对,在SCVs中发现一共有16 946个SNP,其中位于CDs区域的非同义突变SNP为4 154个,同义突变SNP为9 854个(图2A);而位于基因间区的SNP为2 938个。金葡菌SCVs中80%以上的SNP位于蛋白编码区。相同的SNP中,有14 008个SNP位于编码蛋白基因的区域,位于基因间区的SNP为2 938个(图2B)。
比较金葡菌SCVs和CP000253.1参考序列前200个错义突变频率相对较高的基因发现,有66个基因是完全相同的。前200个同义突变密度较高的基因中,有47个是完全相同的。然而,在同一个菌株中,前200个错义突变显著的基因和前200个同义突变显著的基因间并没有重叠。
A.处理前碱基含量分布;B.处理后碱基含量分布A.Base content distribution before processing ;B.Base content distribution after processing
2.4 相关基因的SNP分析
单核苷酸多态性(SNP)主要是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性,包括单个碱基的转换、颠换等。如表3中显示,与CP000253.1参考序列基因组相比,金葡菌SCVs中和耐药能力相关的氨基糖苷类耐药基因的操纵子基因发生了明显的序列变化,该操纵子的aac(6′)、aph(2″)、aph(3′)-Ⅲ基因发生较高频率的错义突变(都属于前200的高错义突变基因)。在金葡菌SCVs菌株中,aph(3′)-Ⅲ的错义突变频率在所有基因中最为显著,约为1.59每100 bp,同义突变频率为0.51每100 bp。

A.金葡菌SCVs菌株在编码蛋白基因区域的分布情况;B.金葡菌SCVs菌株在不同基因组功能区域的分布情况;#Non_syn.位于CDS区域,非同义突变的SNP个数;#Syn.位于CDS区域,非义突变的SNP个数;#Intergenic.位于基因间区的SNP个数
编码β-内酞胺类耐药基因的主要基因mecA的错义突变频率为0.40,qacA基因只发生了同义突变。金葡菌SCVs中另外一些与耐药能力相关的基因aac(6′)和aph(2″) 也发生了显著的错义突变。进一步分析耐药相关基因的转录因子基因ermB、nucA和ypbH序列变化情况,均未发现显著的错义改变(表1) 。
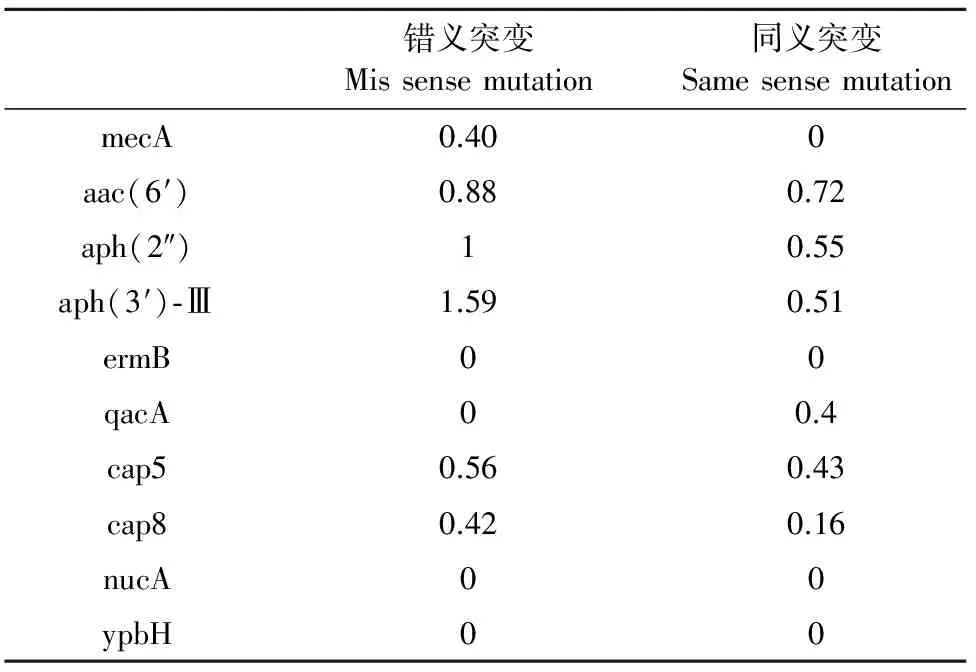
表1 金葡菌SCVs耐药相关基因的SNP频率(每100 bp)
通过采用SAMTOOLS进行个体SNP的检测,全基因组及编码区的转换(transitions)和颠换(transversions)比例、杂合比率及SNP数目的统计结果是,与CP000253.1的参考序列进行比对,SNP检测结果中,SCVs中转换(ts)SNP为10 968个,颠换(tv)SNP为5 978个,SNP的转换颠换率为1.83。所有的16 946个SNP中,杂合(Het)SNP为308,占参考序列基因组的0.01%,纯合(Hom)SNP为16 638个,SNP在参考序列基因组上的密度(density)为6.01/Kb。
3 讨论
通过对金葡菌SCVs菌株的全基因组测序分析,表明金葡菌SCVs菌其基因组序列与实验室菌株CP000253.1相比有很大的差异。研究发现,与CP000253.1菌株相比,野生的金葡菌SCVs菌株在耐药活性、生物膜形成[15]方面都有明显不同。金葡菌SCVs相对于CP000253.1的基因突变率较高,尤其是错义突变基因,暗示在基因组的水平上野生型的菌株相对于驯化的菌株有一些差异,这些差异可能导致了其功能上的差异。另外,跟驯化菌株相比,其耐药的相关基因有十分明显的序列的改变,这些改变可能影响蛋白质的功能,进而有可能影响野生型菌株的耐药能力。在自然的条件下,野生的菌株很难进行转化,很多研究致力于提高野生菌株的转化能力,比较野生型菌株与驯化菌株基因序列上的变化能为更好理解金葡菌SCVs菌株转化能力提供了一种新的途径[1]。同时,这些基因组的差异数据为后续研究金葡菌SCVs的相关物质能力,提高奶牛乳房炎的治疗方面的应用提供更多的数据支持。参考文献:
[1] 王 林,梅 力,韦海涛,等.北京地区奶牛隐性乳房炎金黄色葡萄球菌的分离鉴定及药敏试验[J].动物医学进展,2015,36(4):124-127.
[2] 杨 峰,王 玲,王旭荣,等.金黄色葡萄球菌性奶牛乳房炎研究的知识图谱分析[J].动物医学进展,2016,37(2):38-44.
[3] Fan L,Bo S,Chen H,et al.Genome sequence ofBacillussubtilissubsp.spizizeniigtP20b,isolated from the Indian ocean[J].J Bacteriol,2011,193(5):1276
[4] 喻 钢,田万红,董思国,等.2株枯草芽胞杆菌全基因组重测序分析[J].药物分析杂志,2014,34(10):1907-1911.
[5] 吴三玲.半野生大豆(Glycine gracilis)基因组深度测序及其进化分析[D].浙江杭州:浙江大学,2013.
[6] 张志斌,邓映明,熊瑶瑶,等.东乡野生稻内生放线菌分离及菌株S123 次级代谢产物分析[J].微生物学通报,2015,42(9):1662-1670.
[7] 王琴翠.多粘类芽胞杆菌SC2的全基因组测序[D].山东济南:山东农业大学,2011.
[8] 何伟明.基于重测序数据的群体SNP位点检测及基因型判断[D].广东广州:华南理工大学,2013.
[9] 程廷才,林 平,金盛凯,等.家蚕黑胸败血芽胞杆菌基因组测序及结构分析和功能注释[J].蚕业科学,2014,40(6):1036-1043.
[10] 韩海格.马铜绿假单胞菌全基因组重测序研究[D].内蒙古呼和浩特:内蒙古农业大学,2014.
[11] 刘 超,苏龙翔,方向群,等.铜绿假单胞菌LCT-PA220菌株全基因组序列测定与分析[J].解放军医学院学报,2014,35(7):738-740.
[12] 李泽锋.一个中国南方野生大豆基因组深度测序及其分析[D].浙江杭州:浙江大学,2012.
[13] 陈 重,李多云,程 航,等.一株肠致病性大肠杆菌的全基因组序列分析[J].中国热带医学,2015,15(5):525-528.
[14] 曹德民.一株糖蜜乙醇高产酵母的全基因组重测序研究[D].广西南宁:广西大学,2014.
[15] 魏 武,丁国徽,王晓婧,等.猪链球菌全基因组序列比较分析[J].科学通报,2016,51(7):808-815.
[16] 韩 笑,刀筱芳,张焕容,等.奶牛隐性乳房炎主要病原菌分离鉴定及耐药性分析[J].动物医学进展,2015,36(7):131-134.
Whole Genome Sequencing Analysis of a Small Colony Mutant Strain ofStaphylococcusaureus
WANG Qi-hui,SHI Yong-qiang,ZHU Yu,ZHANG Sai-fei,ZHU Li-li,QU Wei-jie
(FacultyofAnimalScienceandTechnology,YunnanAgriculturalUniversity,Kunming,Yunnan,650201,China)
To study on the genome difference between the small colony mutant strains ofStaphylococcusaureus(SASCVs) and the CP000253.1 for providing data to support subsequent research on the capacity ofS.aureusbacteria secrete of drug related materials.We did a research on a whole genome sequencing of the small colony mutant strains ofStaphylococcusaureus,and compared with those of sequence of CP000253.1 reference sequence.The results revealed that the library into fragments of the small colony mutant strains ofStaphylococcusaureusis 350 bp.The sequencing read long is 150 bp.The amount of raw data of SCVs is 551 Mb.The base content of SCVs distribution is different before and after processing.By sequencing weight,the number of SNP is larger.There is an obvious sequence difference between the small colony mutant strains ofStaphylococcusaureusand the CP000253.1 genome.The data provided the basis in a certain sequence for the study of dairy cow mastitisS.aureusinfection.
S.aureussmall colony mutant strain; difference of gene sequence; whole genome sequencing; SNP
2016-05-13
国家自然科学基金项目(31260629)
王奇惠(1990-),女,山东烟台人,硕士,主要从事动物营养代谢病研究。*通讯作者
S852.611
A
1007-5038(2017)05-0077-04
