高效液相色谱-四极杆/离子阱质谱确证测定面粉及面制品中氨基脲
2017-04-27王翠苹闫革彬
黎 娟,曹 民,王翠苹,闫革彬
(北京市昌平区疾病预防控制中心,北京 102200)
研究简报
高效液相色谱-四极杆/离子阱质谱确证测定面粉及面制品中氨基脲
黎 娟*,曹 民,王翠苹,闫革彬
(北京市昌平区疾病预防控制中心,北京 102200)
采用高效液相色谱-四极杆/离子阱质谱(HPLC-QTRAP-MS)建立了面粉及面制品中氨基脲的确证及测定方法。样品采用盐酸(HCl)提取,在超声辅助下与衍生剂邻硝基苯甲醛反应。衍生产物在中性条件下经PLS固相萃取柱净化、乙酸乙酯洗脱,经Shim-Pack XR-ODS Ⅲ C18柱(2.0 mm×50 mm,1.6 μm)分离,0.1%(体积比)甲酸-水溶液和甲醇溶液为流动相梯度洗脱;采用多反应监测(MRM)-信息依赖性采集(IDA)-增强子离子扫描(EPI)模式检测,EPI谱库确认,内标法定量。结果表明:氨基脲在0.5~40 μg/L范围内线性关系良好(r=0.996);检出限(LOD,S/N=3)为0.10 μg/kg,定量下限(LOQ,S/N=10)为0.25 μg/kg;4个加标水平(0.25,0.5,2.0,10.0 μg/kg)下的回收率为89.1%~112.8%;相对标准偏差(RSD)为1.4%~8.6%。该方法分析速度快,灵敏度高,回收率好,可用于面粉及面制品中氨基脲的快速检测。
高效液相色谱-四极杆/离子阱质谱;氨基脲;超声辅助衍生;面粉及面制品
氨基脲(Semicarbazide,SEM),化学分子式为CH5N3O,是呋喃西林的特征性代谢产物,也是偶氮二甲酰胺在高温条件下的分解产物,可在动物源食品、面制食品等多种食品中被检出[1]。面粉及其制品中的氨基脲主要来源于添加剂偶氮甲酰胺。偶氮甲酰胺是一种能强化面筋、改善面制品组织结构的增筋剂,其在湿润的条件下能够转化为联二脲和脲唑,其中联二脲分子结构与氨基脲相似,可在酸性加热条件下生成氨基脲残留于面粉制品中[2-4]。
目前国内外对于偶氮甲酰胺是否可以作为食品添加剂直接用在面制品中存在不同规定,欧盟、澳大利亚、新加坡等地区和国家已禁止将偶氮甲酰胺作为食品添加剂用于面粉的加工,而在我国偶氮甲酰胺仍可作为添加剂用于面制食品,且在GB 2760-2014《食品添加剂使用卫生标准》中规定其使用上限标准为45 mg/kg[5-6]。有关资料显示,面制品中偶氮甲酰胺与氨基脲的转化效率约为1%,并指出面粉中偶氮甲酰胺的添加量与其转化生成的氨基脲的残留量成正比[7-8]。以上研究暗示若面制品中偶氮甲酰胺的用量为45 mg/kg,则可产生的氨基脲残留量约为450 μg/kg,结合相关毒理学研究结果,即氨基脲具有神经毒性、遗传毒性、生殖毒性及抗雌激素活性等生物毒性效应[9-12],不难推测一旦氨基脲随着食物进入人体,将会对人体健康造成潜在危害。
由于偶氮甲酰胺引起的食品安全隐患归根结底是由于其分解产物——氨基脲具有多种生物毒性效应,因此确证食品中氨基脲的残留水平是监测偶氮甲酰胺引起的健康风险的关键步骤。然而,迄今我国对于面粉及面制品中氨基脲残留量的检测仍无相应的标准。目前主要采用液相色谱-串联质谱法进行检测,由于氨基脲的分子量小、热稳定性差,因此需要衍生后测定。国际上通用的动物组织、面粉及面制品中氨基脲的衍生方法是样品经2-硝基苯甲醛(2-NBA)在恒温振荡下水解衍生16 h。近来尹等[13]建立了超声波辅助衍生,HPLC-MS/MS测定养殖水体中包括氨基脲在内的4种硝基呋喃代谢物的分析方法,超声辅助方法将衍生时间从传统的16 h缩短到60 min,但该报道未考察温度对衍生效率的影响。四极杆/离子阱质谱仪(QTRAP-MS)由于兼具串联四极杆和离子阱的功能,其多反应监测(MRM)-信息依赖性采集(IDA)-增强子离子扫描(EPI)的采集模式可以实现一次进样完成确证和定量分析。因此,近年来应用QTRAP-MS进行检测的文献报道逐渐增加[14-16],但使用QTRAP-MS检测氨基脲的文献尚无报道。本文基于高效液相色谱-离子阱质谱,即应用QTRAP-MS,采集模式MRM-IDA-EPI,结合优化超声衍生温度、时间等前处理条件及色谱-质谱条件,建立了面粉及面制品中氨基脲确证和测定的分析技术。
1 实验部分
1.1 仪器与试剂
API 4500 QTRAP型三重四极杆串联质谱仪配电喷雾离子源(美国AB 公司);LC-30A型高效液相色谱(日本岛津公司);2-16KL型冷冻离心机(美国Sigma公司);GL Sciences固相萃取装置(Dikma公司);氮吹仪(美国Organomation 公司);旋涡震荡仪(海门市麒麟医用仪器厂)。
甲醇、甲酸(色谱纯,Dikma公司);氢氧化钠(分析纯,国药集团化学试剂有限公司);浓盐酸(优质纯,国药集团化学试剂有限公司);邻硝基苯甲醛(Alfa Aesar公司,纯度98%);氨基脲盐酸盐(SEM-HCl,Fluka公司,纯度99%);氨基脲盐酸盐同位素内标(SEM-[1,2-15N213C],Fluka公司,纯度99%)。PLS固相萃取柱(Dikma公司)。
1.2 液相色谱条件
色谱柱:Shim-Pack XR-ODS Ⅲ C18(2.0 mm×50 mm, 1.6 μm);流动相:A为0.1%甲酸水溶液,B为甲醇。流速:0.3 mL/min;进样量:5 μL;梯度洗脱程序:0~0.1 min,10%B;0.1~6 min,10%~30% B;6~8 min,30%~90% B;8~10 min,90% B;10~12 min,10% B。柱温箱:40 ℃;分析时间12 min。
1.3 质谱条件
用正离子模式(ESI+),在MRM-IDA-EPI扫描模式下采集数据;MRM检测窗口:60 s;目标扫描时间:1 s;IDA采集阈值:500 cps(勾选动态背景扣除);EPI扫描速率:10 000 Da/s;扫描质量数范围:50~300。优化后的质谱分析条件:离子化电压(IS):5 500 V;离子源温度(TEM):550 ℃;气帘气压力(CUR):1.7×105Pa;喷雾气(GS1):3.8×105Pa;辅助加热气(GS2):4.0×105Pa。定量离子、定性离子、去簇电压(DP)、碰撞能量(CE)见表1。

表1 氨基脲及其同位素内标的质谱条件Table 1 MS parameters for the determination of SEM and SEM isotopes
*quantitative ion;DP:declustering potential;CE:collision energe
1.4 样品处理方法
1.4.1 样品水解与衍生化 称取混匀的试样 2 g(准确至0.001 g),置50 mL 塑料离心管中,加氨基脲同位素内标溶液(0.1 μg/mL) 50 μL和盐酸(0.2 mol/L)10 mL,在涡旋混合器上混合均匀后,加入100 μL 0.1 mol/L 2-硝基苯甲醛溶液,再次混匀后,置于超声波仪中在37 ℃下避光超声30 min。
1.4.2 样品的提取与净化 将衍生化后的试样冷却至室温,加NaOH溶液(2.0 mol/L)1.1 mL调节pH 值(6.8~7.2),于4 ℃下,10 000 r/min 离心10 min,取上清液,待过PLS柱。依次用3 mL甲醇和3 mL水活化PLS小柱,上样,以6 mL水淋洗。淋洗后吹干,加乙酸乙酯3 mL洗脱。洗脱液在40 ℃下氮吹至近干,残渣以甲酸溶液(0.1%)1 mL溶解,离心(10 000 r/min,10 min,4 ℃),取上清液检测。
1.5 空白基质匹配标准溶液的配制
使用不含有氨基脲的面粉作为空白基质,称取2 g(准确至0.001 g)置于50 mL 塑料离心管中,加盐酸(0.2 mol/L) 10 mL,在涡旋混合器上混合均匀。分别加氨基脲标准溶液(0.020 μg/mL) 0,25,50,100,250,500,2 000 μL,并分别加氨基脲同位素内标溶液(0.10 μg/mL) 50 μL。其余操作按照“1.4.1”和“1.4.2”所述。
2 结果与讨论
2.1 流动相的选择

食品中氨基脲的HPLC-MS/MS检测方法中,关于流动相的选择有所不同:如农业部781号公告中采用5 mmol/L甲酸铵-0.1%甲酸作为流动相分析动物源食品中包括氨基脲在内的多种硝基呋喃代谢产物;而王丹等[16]经过与农业部781号公告中的方法比较,认为以0.1%的甲酸水和纯乙腈溶液作为流动相可降低质谱的背景信号值、提高信噪比。本实验以0.1%的甲酸水作为流动相A,并比较了以纯甲醇和纯乙腈分别作为流动相B的情况下目标化合物峰的分离程度,发现选择纯乙腈为流动相B时氨基脲的保留时间为4.25 min,而选用纯甲醇为流动相B时,氨基脲的保留时间为6.22 min,除了保留时间有所差别,其余无任何影响。考虑到乙腈成本高,且毒性大,所以本实验选择甲醇作为流动相B,以达到较好的色谱分离目的,同时又可降低实验成本和提高安全操作保障。图1为本实验条件下HPLC-MS/MS分析氨基脲及其同位素内标的MRM色谱图。
2.2 样品衍生化条件的选择
HPLC-MS/MS检测食品中的氨基脲,一般采用2-NBA衍生氨基脲,形成特征性衍生物,以提高质谱检测的灵敏度。超声波的高频振荡可加速样品分散,使得样品与溶剂的接触面积增大,进而提高传质速率,因此使用超声辅助衍生具有节约衍生反应时间的优势。本研究采用内标定量法,以回收率为指标,选择37,45,60 ℃ 3个衍生温度梯度,分别在15,30,60,120,180,240 min超声时间下进行衍生化反应。结果显示,衍生温度为37 ℃和45 ℃时,上述6个超声时间段下的回收率分别为103.7%,103.4%,103.8%,103.5%,103.6%,103.9%和104.1%,104.4%,104.8%,104.4%,104.8%,103.2%;而衍生温度为60 ℃时,各超声时间段下的回收率分别为109.8%,108.2%,107.8%,106.9%,107.6%及109.6%。超声衍生时间的比较实验结果显示,对于同一衍生温度,其回收率在各超声时间段下无明显差异。初步优化结果表明衍生温度对衍生效率有一定影响,37 ℃或45 ℃可作为适宜衍生温度采用,而超声衍生时间对衍生效率无明显影响,即超声时间段从15 min至240 min均可。
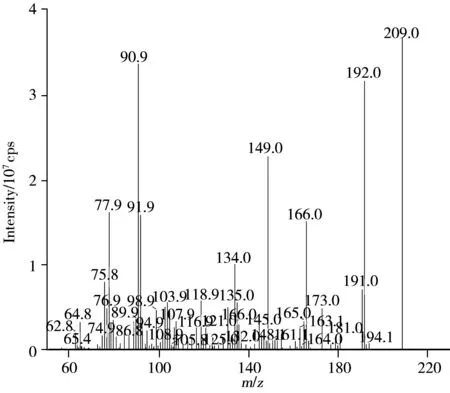
图2 1 μg/L氨基脲标准溶液的在线EPI谱图Fig.2 On-line EPI spectrum of 1 μg/L SEM standard solution
2.3 质谱条件的选择
采用1 μg/L氨基脲标准溶液进行质谱参数优化,以MRM-IDA-EPI模式采集氨基脲的EPI谱图(图2),所得到的质谱相关参数如“1.3”所述。与MRM扫描模式相比,MRM-IDA-EPI采集模式同时具有筛查、定量和确证功能,能够获得较为丰富的确证信息。本方法中EPI模式是在3种采集能量(20,35,50 V)水平上获得离子碎片的数学组合,即一次采集可获得3种能量级(20,35,50 V)的EPI图谱。如图2所示,EPI谱图中除了包含MRM采集的m/z209,166外,还有m/z192,177,149,64.8等碎片离子的信息,根据欧盟关于鉴别点(IdentificationPoint,IP)的计算规则[17],母离子IP为1.0,子离子IP为1.5,使用该采集模式获得的氨基脲IP值为8.5,远高于欧盟规定的污染物确认方法中IP值应为4.0的要求。由于离子阱的灵敏度高于四极杆,因此即使是低浓度水平的检测,EPI谱图也可获得较好的确证优势。
2.4 定量下限及线性关系
取实际加标样品溶液,用0.1%甲酸水倍比稀释后测定,以信噪比(S/N)为3时所测定实际加标样品溶液中氨基脲的浓度为检出限,以S/N为10时氨基脲的浓度为定量下限。在标准溶液中,以目标组分定量离子的相对峰面积比值(y)对相应的质量浓度(x,μg/L)绘制标准曲线(浓度范围:0.5,1.0,2.0,5.0,10,40 μg/L),线性回归采用1/x2权重(校正低浓度端准确度),所得线性回归方程为y=2.27x+0.003 98,线性关系良好(r=0.996)。当取样量为2 g时,本研究中氨基脲的检出限为0.10 μg/kg,定量下限为0.25 μg/kg。运用LC-MS/MS检测面粉中的氨基脲,周、刘等得到的检出限分别为0.15,0.25 μg/kg[6,18],而王、周等获得的定量下限为0.50 μg/kg[16,18],均达到国际要求的检出限0.50 μg/kg。可见,与上述报道的数据相比,本方法可以获得更佳的检出限与定量下限。
2.5 回收率与精密度
以阴性面粉为样品基质,进行4个浓度水平(0.25,0.5,2.0,10.0 μg/kg)的加标回收检测,每个浓度水平进行6次平行实验,内标法定量,计算其平均回收率及相对标准偏差(RSD)。结果显示,在4个加标水平下氨基脲的平均回收率范围分别为89.1%~112.8%,95.6%~109.6%,101.3%~108.8%,97.9%~102.3%,RSDs范围分别为6.9%~8.6%,4.9%~5.6%,3.6%~3.8%及1.4%~1.5%。
2.6 EPI谱图匹配纯度阈值的确定及运用
采用LC-MS/MS分析物质残留时,通常将鉴别点数(IPs)、保留时间(RT)及信噪比(S/N)等作为物质定性的参数。本方法使用EPI图谱比对辅助进行面粉及其制品中氨基脲残留量确证:用IDA触发保留时间匹配(±2.5%)且信噪比满足要求(S/N≥3)的质量色谱峰,采集其EPI谱图,进一步与谱库中目标物的标准EPI谱图进行比对,使用Analyst软件计算EPI谱图匹配的Fit、RevFit及Purity值。
Purity阈值确定方法参见文献[15,19],即通过空白加标实验与标准溶液相比对确定,取阴性面粉样品作为空白基质加标(以定量下限0.25 μg/kg为加标量),测定10个平行试样,采集氨基脲的EPI谱图与标准EPI库进行比对。计算结果显示,空白基质添加样品中的EPI谱图对标准工作液和对基质标准工作液的EPI谱图匹配Purity值无明显差别,故本实验以对标准工作液的Purity值70.44(±2.88)为氨基脲的EPI谱图纯度阈值,样品中氨基脲的EPI谱图匹配Purity值超过70.44即可作为确证分析的有效判据。
为了进一步确认最佳衍生时间和温度,本研究根据建立好的高效液相色谱-四极杆/离子阱质谱条件,并结合“2.2”所述,分别对比分析37,45,60 ℃下,超声衍生时间为15 min和30 min的氨基脲EPI谱图,以EPI谱图比对的匹配结果作为辅助指标判断最优衍生条件。结果表明,综合考虑匹配结果的Fit,RevFit及Purity值,则最佳衍生温度和时间分别为37 ℃和30 min(表2);在实际样品检测中均采用该衍生条件,即在37 ℃下超声衍生30 min。
2.7 实际样品的检测
利用本方法对53件北京市售的面粉及其制品(面包、蛋糕、饼干、包子)中的氨基脲进行检测,检出阳性样品24件,含量为0.171~913 μg/kg。图3为1个检出量为244 μg/kg的面包样品的相关图谱。其相应目标峰EPI图谱匹配在线谱库中标准的氨基脲EPI图谱的Fit,RevFit及purity值分别88.41,92.28及87.09,可有效确证面包中氨基脲的存在。



图3 面包样品中氨基脲的选择离子流质量色谱图(A)、在线EPI谱图(B)及谱库中氨基脲的标准EPI谱图(C)Fig.3 Extracted ion chromatogram of SEM in positive bread sample(A),on-line EPI spectrum of SEM(m/z 209.1/166.0) in positive bread sample(B) and on-line EPI matched spectrum of SEM in library(C)
3 结 论
本文基于高效液相色谱-四极杆/离子阱质谱(HPLC-Q/Trap-MS)建立了面粉及面制品中氨基脲的确证及测定方法。在样品提取中,采用超声波辅助衍生方法代替传统的恒温振荡法,并在此基础上探讨了超声波辅助衍生的最佳温度和时间,发现超声温度为37 ℃、衍生时间为30 min时衍生效率达到最佳。此外,本方法采用多反应监测(MRM)-信息依赖性采集(IDA)-增强子离子扫描(EPI)模式检测可得到较高的IP值,且可同时得到3个能量级的EPI图谱,通过将其与谱库中标准EPI图谱对比,实现了对氨基脲的确证和定量分析。该方法分析速度快,灵敏度高,回收率好,可有效用于面粉及其制品中氨基脲的快速检测,并为面粉及其制品中氨基脲的监管提供了强有力的技术支撑。
[1] Tian W R,Sang Y X,Wang X H.Food.Addit.Contam.,2014,31(11):1850-1860.
[2] Noonan G O,Begley T H,Diachenko G W.J.Agric.FoodChem.,2008,56(6):2064-2067.
[3] Wang Y,Wang J S,Xiang L,Xi C X,Chen D D,Peng T,Wang G M,Mu Z D.Chin.J.Chromatogr.(王雅,王俊苏,向露,郗存显,陈冬东,彭涛,王国民,母昭德.色谱),2014,32(5):513-518.
[4] Zhang J H,Ouyang L,Peng X K,Xia L X,Li H,Wang H.J.Instrum.Anal.(章建辉,欧阳丽,彭新凯,夏立新,李欢,汪辉.分析测试学报),2015,34(12):1430-1433.
[5] GB 2760-2011.Standards for Using Food Additives.National Standards of Food Safety(食品添加剂使用标准.食品安全国家标准).
[6] Zhou Q M,Xiang L,Wang J S,Xi C X,Wang Y,Peng T,Mu Z D,Wang G M.Chin.J.Anal.Lab.(周启明,向露,王俊苏,郗存显,王雅,彭涛,母昭德,王国民.分析试验室),2014,(7):782-786.
[7] Noonan G O,Warner C R,Hsu W,Begley T H,Perfetti G A,Diachenko G W.J.Agric.FoodChem.,2005,53(12):4680-4685.
[8] Jiang Z H,Wu X P,Wang M X,Hong P Z,Chen H J,Lai Z,Chen Y L.FoodSci.(蒋志红,吴晓萍,王明兴,洪鹏志,陈华健,赖周,陈煜玲.食品科学),2014,35(19):91-95.
[9] Maranghi F,Tassinari R,Lagatta V,Moracci G,Macrì C,Eusepi A,Di Virgilio A,Scattoni M,Calamandrei G.FoodChem.Toxicol.,2009,47(2):472-479.
[10] Maranghi F,Tassinari R,Marcoccia D,Altieri I.Catone T,De Angelis G,Testai E,Mastrangelo S,Evandri M G,Bolle P.Chem.Biol.Interact.,2010,183(1):40-48.
[11] Vlastos D,Moshou H,Epeoglou K.FoodChem.Toxicol.,2010,48(1):209-214.
[12] Gao S,Ru S G.Res.Environ.Sci.(高素,汝少国.环境科学研究),2013,26(6):637-644.
[13] Yin Y,Pan Y,Liu S G,Zheng G M,Zhu X P,Ma L S,Dai X X,Shan Q.Chin.J.Anal.Lab.(尹怡,潘宇,刘书贵,郑光明,朱新平,马丽莎,戴晓欣,单奇.分析试验室),2015,4:416-420.
[14] Chen Y,Zhao L,Lu F,Yu Y,Chai Y,Wu Y.Food.Addit.Contam.,2009,26(5):595-603.
[15] Zhang H W,Jian H M,Lin L M,Chen L Z,Liang C Z,Yuan T,Tang Z X,Cai X,Qin L Y.J.Instrum.Anal.(张鸿伟,简慧敏,林黎明,陈亮珍,梁成珠,袁涛,汤志旭,蔡雪,秦良勇.分析测试学报),2012,31(7):763-770.
[16] Wang D,Chen Y,Song S F,Gao J,Liang J,Zhao Y F.Chin.J.FoodHyg.(王丹,陈颖,宋书锋,高洁,梁江,赵云峰.中国食品卫生杂志),2014,26(6):579-583.
[17] Commission E:Commission Decision EC 2002/657 of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results.Off.J.Eur.Communities L 2002,221.
[18] Liu Q F.AnhuiChem.Ind.(刘琴芳.安徽化工),2013,39(5):79-81.
[19] Zhang H W,Cai X,Lin L M,Chen L Z,Liang C Z,Bao L,Tang Z X,Niu Z Y,Wang F M.Chin.J.Chromatogr.(张鸿伟,蔡雪,林黎明,陈亮珍,梁成珠,鲍蕾,汤志旭,牛增元,王凤美.色谱),2012,30(10):991-1001.
Identification and Detection of Semicarbazide in Flour and Flour Products by High Performance Liquid Chromatography Coupled to Quadrupole/Linear Ion Trap Mass Spectrometry
LI Juan*,CAO Min,WANG Cui-ping,YAN Ge-bin
(Changping Center for Disease Control and Prevention,Beijing 102200,China)
An analytical method was developed for the identification and detection of semicarbazide(SEM) residues in flour and flour products by high performance liquid chromatography coupled to quadrupole/linear ion trap mass spectrometry(HPLC-QTRAP/MS).Samples were reacted with o-nitrobenzaldehyde in HCl with assistant of ultrasonic to produce stable derivative,and then the derivative was cleaned up with PLS SPE cartridge,eluted with ethyl acetate.The separation of analytes was carried out on a Shim-pack XR-ODS Ⅲ C18column(2.0 mm×50 mm,1.6 μm) using mobile phases of 0.1%(by volume) formic acid in ultrapure water and methanol solution by gradient elution.A multiple reaction monitoring(MRM) scan mode and an enhanced product ion(EPI) scan mode as dependent in an information-dependent acquisition(IDA) experiment were used in mass spectrometry acquisition.Lab-built EPI library and the internal standards were adopted for the identification and quantification.The results indicated that the calibration curve of SEM showed a good linearity(r=0.996) whithin the range of 0.5-40 μg/L.The limit of detection(LOD,S/N=3) for SEM was 0.10 μg/kg,and the limit of quantitation(LOQ,S/N=10) was 0.25 μg/kg.The average recoveries of SEM at four spiked levels of 0.25,0.5,2.0,10.0 μg/kg were in the range of 89.1%-112.8% with relative standard deviations(RSDs) of 1.4%-8.6%.With high sensitivity and good recovery,the proposed method was effectively used to identify and detect the residues of semicarbazide in flour and flour products.
high performance liquid chromatography-quadruple/linear ion trap mass spectrometry(HPLC-QTRAP/MS);semicarbazide;ultrasonic-assisted derivatization;flour and flour products
10.3969/j.issn.1004-4957.2017.04.008
2016-11-14;
2016-12-10
中国科学院生态环境研究中心环境化学与生态毒理学国家重点实验室开放基金(KF2015-04)
O657.63;TS213.2
A
1004-4957(2017)04-0490-06
*通讯作者:黎 娟,博士,主管技师,研究方向:食品科学与毒理学,Tel:010-69720125,E-mail:allison_2008@126.com
