固相萃取-气相色谱法检测血清中有机氯农药残留的研究
2017-01-06段丽村周建于李佳欣李鹏飞
段丽村,周建于,李佳欣,李鹏飞,徐 芳,王 琦
(昆明医科大学 公共卫生学院,云南 昆明 650500)
固相萃取-气相色谱法检测血清中有机氯农药残留的研究
段丽村,周建于,李佳欣,李鹏飞,徐 芳,王 琦*
(昆明医科大学 公共卫生学院,云南 昆明 650500)
建立了血清中DDTs和BHCs共8种有机氯农药残留的固相萃取-气相色谱检测方法。样品经超声酸化沉淀蛋白后,采用正己烷-丙酮(9∶1)经Cleanert ODS C18N固相萃取小柱提取,Florisil固相萃取小柱净化,氮气吹干,以500 μL正己烷定容,气相色谱-电子捕获检测器(GC-ECD)进行定量分析。结果表明,方法的线性范围2~200 ng/mL,相关系数(r)为0.996 4~0.999 0,检出限(LOD) 为0.1~0.9 ng/mL,定量下限(LOQ)为0.4~3.0 ng/mL。8种农药的回收率为80.5%~112.7%,相对标准偏差(RSD)为2.1%~7.9%。该方法具有较高的准确度和精密度,适用于血清样品中痕量有机氯农药的检测。
血清;DDTs;BHCs;有机氯农药;固相萃取;气相色谱
有机氯农药(OCPs) 具有高效、低成本、广谱杀虫等特点,在我国20世纪50年代之后大量使用。因其污染严重,在环境中的长期残留性、生物蓄积性和高毒性等特征[1],我国于1983年停产使用六六六、滴滴涕,但在很多曾施用有机氯农药的地方仍有大量残留[2]。已有研究表明,DDT已进入全球性的生物地球化学循环,成为世界各国关注的环境和健康问题[3]。截至2009年,已有约十余种有机氯农药被《关于持久性有机污染物的斯德哥尔摩公约》列为典型的持久性污染物(Persistent organic pollutants,POPs)及优先控制有机污染物加以控制[4-5]。
有机氯及其代谢产物的化学性质稳定、脂溶性强、难降解,目前在环境和生物样品中仍然可以检测到有机氯农药[6-7]。该类化合物可通过消化道、呼吸道和皮肤吸收,分布到各组织和器官,在血液中几乎全部与血浆蛋白结合[8-9]。因此,对生物样品中有机氯农药进行分析检测是了解其在生物体内暴露的基础。对于一般人群的暴露研究,血液是较易获得的样本。由于有机氯及其代谢产物在人体血液中的含量大致与其接触量成正比关系,因此,检测人体血液中的有机氯含量可以反映人群暴露于污染物的水平。
血清中有机氯农药残留的分析,国外通常采用固相萃取法进行提取和净化,GC-MS检测,且常与其它卤代有机物如多氯联苯(PCBs)、多溴联苯醚(PBDEs)等同时检测[10];而六六六(Hexachlorocyclohexane,BHC) 和DDT(Dichlorodiphenyltrichloroethane) 的测定大多采用电子捕获检测器检测的气相色谱法,国内对该方面的研究较少,一般用液液萃取提取待测物,再用浓硫酸或浓硫酸与Florisil柱联用净化,气相色谱-电子捕获检测器(GC-ECD)分析定量[11]。电子捕获检测器虽在定性能力上弱于质谱,但对于有机氯的检测灵敏度与MS相当,也是分析含氯化合物的常用检测器。本研究以人血清中BHC 4种异构体和DDT及其主要代谢产物为研究对象,建立了一种快速、准确、灵敏的检测方法,为人群有机氯暴露水平调查以及评价有机氯农药污染与人群健康的关系提供了可靠的检测手段。
1 实验部分
1.1 仪器与试剂
气相色谱仪配电子捕获检测器(ECD,Varian CP-3800美国瓦里安公司);Rtx-5毛细管色谱柱(30 m×0.25 mm×0.25 μm,美国Restek公司);DG-12D固相萃取装置(12孔,上海皓庄仪器有限公司);WH-1微型涡旋混合仪(上海泸西分析仪器厂);KQ3200超声波清洗仪(科桥超声波设备有限公司);氮吹仪(上海济成分析仪器公司)。
α-BHC,β-BHC,γ-BHC,δ-BHC,p,p′-DDE,p,p′-DDD,o,p′-DDT,p,p′-DDT 8种农药标准品(农业部环境保护科研监测所);甲醇、二氯甲烷、甲酸(分析纯,北京化学试剂厂);无水硫酸钠(分析纯,天津化学试剂厂);正己烷(色谱纯,德国Merk公司);高纯N2(纯度>99.999%,昆明梅塞尔气体有限公司);ODS C18N柱、Florisil柱(均为500 mg/3 mL,天津博纳艾杰尔公司);Silica柱(500 mg/3 mL,北京迪马科技有限公司)。
1.2 溶液的配制
8种有机氯农药标准品的初始浓度为100 μg/mL,用正己烷配成10 μg/mL的混标储备液,使用前稀释至1.00 μg/mL,贮存于4 ℃冰箱。无水硫酸钠使用前在马弗炉中450 ℃灼烧4 h。
1.3 色谱条件
进样口温度250 ℃,检测器温度310 ℃,载气为高纯氮气,流速1 mL/min,尾吹30 mL/min,不分流模式进样量1 μL。毛细管色谱柱升温程序:100 ℃保持1 min,以10 ℃/min升至200 ℃,保持2 min,再以 4 ℃/min升至250 ℃,保持1 min,共26.5 min。
1.4 样品采集与制备
选取昆明市呈贡区雨花村和乌龙村50~70岁男性居民,抽取空腹静脉血,静置30 min,3 500 r/min离心10 min,取上层血清,放入-80 ℃冰箱保存备用。预实验选用混合血清进行。
1.5 样品前处理过程
1.5.1 沉淀蛋白 血清样品室温解冻,取1 mL于10 mL离心管中,加纯水1 mL,涡旋2 min混匀,加入甲酸0.5 mL并涡旋2 min,超声20 min酸化沉淀蛋白。
1.5.2 提 取 Cleanert ODS C18N固相萃取小柱的活化过程:先用5 mL二氯甲烷淋洗,再加入8 mL甲醇淋洗,8 mL纯水平衡,保持小柱不干。将沉淀蛋白质的血清样品加入活化好的C18柱,用10 mL纯水冲洗去除杂质,抽干柱中水分并在空气中晾干30 min,待测物用10 mL正己烷-丙酮(9∶1)洗脱,控制流速为1~2 mL/min。
1.5.3 净 化 将10 mL正己烷-丙酮(9∶1)洗脱液用氮气吹干至3 mL,过装有2 g无水Na2SO4的小漏斗并用1 mL正己烷冲洗。Florisil固相萃取小柱活化:加入8 mL正己烷-丙酮(9∶1)溶液冲洗柱子,保持小柱不干,将上述脱水的溶液加入已活化的小柱,收集流出液,并用6 mL正己烷-丙酮(9∶1)溶液淋洗小柱,合并滤液后,氮气吹干溶液,用500 μL正己烷溶解,过0.45 μm滤膜,待GC检测。
1.6 二甲基氯硅烷(DMCs)对衬管硅烷化处理
采用丙酮等有机溶剂将衬管清洗干净、晾干后,用5% DMCs正己烷溶液浸泡过夜,取出用甲醇清洗2~3次,浸泡1 h后,取出晾干,干燥条件下保存备用。
2 结果与讨论
本研究选取不含目标待测物的混合血清样品,每1 mL样品加入25 μL浓度为1 μg/mL的8种有机氯农药混标,重复3次,优化样品前处理条件。
2.1 色谱条件的优化
2.1.1 色谱柱的选择 有机氯农药属于弱极性至中等极性的化合物,本实验采用中等极性的Rtx-1701和Rtx-5两种毛细管色谱柱对待测物进行分离,发现在不同色谱条件下,两种色谱柱均能较好地分离8种待测有机氯农药。根据实验室现有条件,采用Rtx-5色谱柱进行分离。
2.1.2 载气流速的选择 载气流量是影响待测物在色谱柱中分离效率的重要因素,载气流量慢,各峰的分离度较好,但所用分析时间长;载气流量较快,分析时间缩短,但分离度变差。实验考察了柱流量为1 mL/min和2 mL/min 对分离效果的影响,发现柱流量增大时,各待测物的保留时间提前,但o,p′-DDT与p,p′-DDD部分色谱峰重叠,不能达到完全分离。故选择柱流量为1 mL/min,在该柱流量下,各待测物分离效果好。
2.1.3 衬管硅烷化处理 由于p,p′-DDT可能会在进样口分解为p,p′-DDE和p,p′-DDD,所以参照美国EPA8081B方法,检测了一定数量的样品后,按“1.6”方法对气相色谱进样口衬管进行清洗和硅烷化处理,更换进样垫,并将毛细管色谱柱连接进样口的一端截除一段。
2.2 沉淀蛋白
人血清中含有的脂肪酸、胆固醇、甘油三脂和蛋白质易附着在色谱柱上,从而改变柱子的特性并降低柱寿命,同时其中的游离脂肪酸可能会干扰色谱测定。根据文献报道,甲酸是沉淀这些脂质和蛋白质的良好沉淀剂,故实验采用甲酸作为沉淀剂。分别取1 mL血清,各加入甲酸0.2,0.5,1 mL沉淀蛋白,按操作步骤进行处理和检测,发现加入甲酸0.5 mL和1 mL时,杂峰明显减少,且效果相当,所以本实验选择加入0.5 mL甲酸沉淀血清中的蛋白质。
2.3 提取条件的优化
2.3.1 固相萃取柱的选择 固相萃取是近年用于液体样品前处理的新技术。当液体中有机化合物通过合适的固相萃取柱时被富集,再用少量选择性溶剂洗脱,因而固相萃取是同时完成萃取和浓缩有机污染物的有效方式[12-14]。该方法操作简单、流程短,同时有机溶剂的用量较少,符合快速、高效、低污染的样品前处理方法[14]。因此本实验选择固相萃取法对血清中有机氯农药进行提取。
C18材料的作用基团是十八烷基,对非极性、弱极性和中等极性化合物具有广泛保留,是目前固相萃取中应用最广的吸附剂。C18-N是硅胶键合C18后未进行封端处理的反相柱,作用基团为十八烷基和硅羟基,对极性化合物的保留增强。根据待测物的理化性质和样品基质的特点,选择对待测物有较强保留能力的C18反相填料作为固定相进行萃取。
2.3.2 洗脱溶剂的选择 由于有机氯农药属于弱极性至中等极性的化合物,实验考察了正己烷、正己烷-二氯甲烷(9∶1)、正己烷-丙酮(9∶1) 3种溶剂体系对待测物的洗脱效果。各取纯水1 mL,按样品前处理步骤,沉淀蛋白,过C18柱,分别用上述3种溶剂15 mL对待测农药进行淋洗。结果显示,正己烷、正己烷-丙酮(9∶1)作为洗脱溶剂时8种有机氯农药的回收率相对较高。
进一步考察了洗脱溶剂正己烷及正己烷-丙酮(9∶1)用量的影响。用12 mL该溶剂进行淋洗,每收集2 mL作为1份,进行洗脱曲线的制作。结果显示,以正己烷作为洗脱溶剂时,需加入15 mL才能将δ-BHC洗脱完全,而正己烷-丙酮(9∶1)只需10 mL则可将8种目标农药洗脱完全,所以实验采用10 mL正己烷-丙酮(9∶1)作为最佳洗脱溶剂。
2.4 净化条件的优化
2.4.1 固相萃取柱的选择 采用“1.5”方法分别对Florisil柱和Si柱进行活化,上样,收集流出液,采用正己烷、正己烷-丙酮(9∶1)、正己烷-二氯甲烷(1∶1)各20 mL进行淋洗,过0.45 μm滤膜,GC检测。结果表明,采用正己烷-丙酮(9∶1)作为洗脱溶剂,Florisil柱与Si柱的净化效果相当,但Si柱净化后杂峰相对较多,考虑到后续将对多个血清样品进行检测,本实验采用Florisil柱对血清样品进行净化。
2.4.2 洗脱溶剂体积的选择 用Florisil柱净化,正己烷-丙酮(9∶1)为洗脱溶剂,对洗脱所需体积进行考察。用6 mL该溶剂进行淋洗,每收集1 mL作为1份,共6份进行洗脱曲线的制作。结果显示,前5 mL洗脱液可以洗脱大部分农药,为确保待测物洗脱完全,实验选择正己烷-丙酮(9∶1)的最佳用量为6 mL,且采用的流速为小柱自然流速,以便洗脱液与待测物充分接触。
2.5 方法评价
2.5.1 线性范围、检出限及定量下限 配制不同浓度梯度的混合标准溶液,在最佳色谱条件下,以峰面积(Y)为纵坐标,质量浓度为横坐标(X,ng/mL)制作标准曲线,分别以3倍信噪比(S/N=3)和10倍信噪比(S/N=10)计算检出限(LOD)和定量下限(LOQ)。结果如表1所示,方法的线性范围2~200 ng/mL,相关系数(r)为0.996 4~0.999 0,LOD为0.1~0.9 ng/mL,LOQ为0.4~3.0 ng/mL。

表1 方法的特征参数
2.5.2 回收率与精密度 在空白血清样品中分别加入5,20 ng/mL两个浓度梯度的有机氯标准品,在最佳条件下进行样品前处理和检测。结果如表2所示,8种农药的回收率为80.5%~112.7%,相对标准偏差(RSD)为2.1%~7.9%。所建立的方法具有较高的准确度和精密度,能用于血清样品中痕量有机氯农药的分析检测。
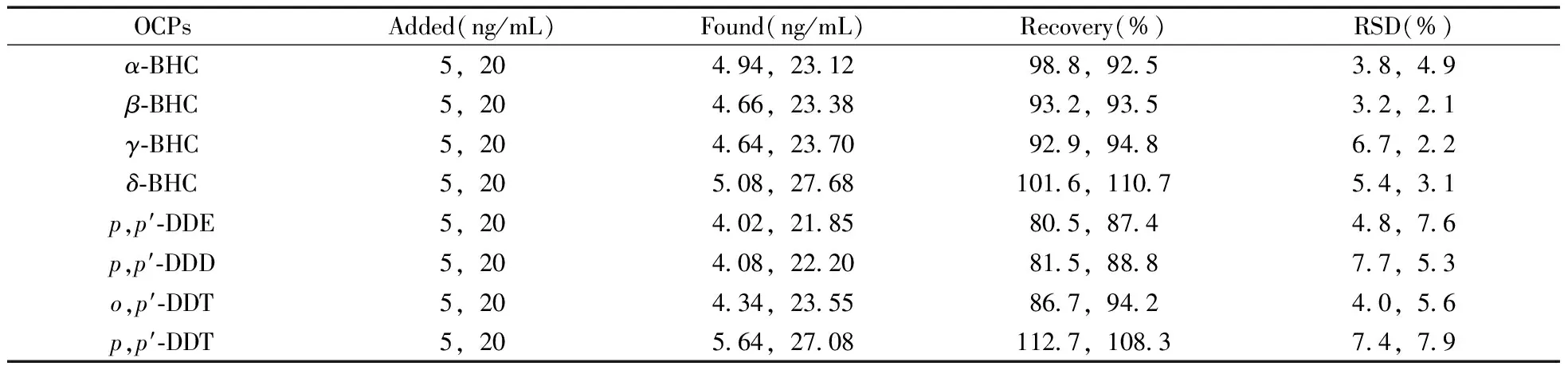
表2 血清样品的加标回收率与相对标准偏差(n=5)
2.5.3 实际样品的分析 将建立的方法用于调查对象血清中8种有机氯农药的检测,图1为5 ng/mL标准溶液和第56号样品的色谱图。对于标准溶液,8种有机氯农药的保留时间见表1。实际样品中检出4种有机氯农药,其中α-BHC为1.87 ng/mL,p,p′-DDE为2.86 ng/mL,p,p′-DDD为2.48 ng/mL,o,p′-DDT为2.60 ng/mL。实验结果表明,利用C18和Florisil固相萃取柱分别对样品进行提取和净化,正己烷-丙酮(9∶1)溶剂对待测物进行洗脱后,8种有机氯农药的精密度和回收率均较为理想。该方法快速、高效,能够满足血清中有机氯农药残留检测的要求。

图1 有机氯农药标准溶液(A,5 ng/mL)及血清样品(B)的色谱图
[1] Yang B,Han B,Xue N,Zhou L L,Li F S.J.Environ.Sci.,2015,27:241-250.
[2] Zhou C,Wang Z C,Luo M B,Yao P P,Yuan Z Z.J.Instrum.Anal.(周纯,王志畅,罗明标,姚培培,袁自遵.分析测试学报),2013,32(11):1354-1358.
[3] Liu G H,Wang Y,Liu H K.Chin.J.Androl.(刘国红,王翀,刘宏凯.中华男科学杂志),2006,12(2):104-107.
[4] Cai Y Z,Wang Y W,Jiang G B.Sci.Sin.:Chim.(蔡亚岐,王亚韡,江桂斌.中国科学:化学),2010,40(2):99-123.
[5] Song S L,Rao Z,Ma X D,Sun W L.J.Instrum.Anal.(宋淑玲,饶竹,马晓东,孙玮琳.分析测试学报),2011,20(1):108-114.
[6] Qiu X H,Zhu T,Yao B,Hu J X,Hu S W.Environ.Sci.Technol.,2005,39(12):4385-4390.
[7] Qi S H,You Y H,Su Q K,Gong X Y,Wu C X,Wang W,Lü C L.Geol.Bull.Chin.(祁士华,游远航,苏秋克,龚香宜,吴辰熙,王伟,吕春玲.地质通报),2005,24(8):704-709.
[8] Yoshida R,Fukami M,Sasagawa I,Hasegawa T,Kamatani N,Ogata T.J.Clin.Endocirnol.Metab.,2005,90(8):4716-4721.
[9] Vogel J M.EnvironHealth,2005,4:1476-1488.
[10] Su J F,Zhong M S,Chen J,Guo X.J.Instrum.Anal.(苏建峰,钟茂生,陈晶,郭昕.分析测试学报),2015,34(6):625-638.
[11] Li Y Q,Zeng H Y,Zou X L,Chen L Q.Mod.Prevent.Med.(黎源倩,曾红燕,邹晓莉,陈路齐.现代预防医学),2007,34(3):407-411.
[12] Wang Y,Zhi X X,Zhang L J.RockMiner.Anal.(汪雨,支辛辛,张玲金.岩矿测试),2006,25(4):493-496.[13] Liu J T.Chin.J.Chromatogr.(刘俊亭.色谱),1997,15(2):118-119.
[14] Chen W Q.EnergyEvniron.(陈武强.能源与环境),2015,(5):29-31.
Determination of Organochlorine Pesticide Residues in Serum by Solid Phase Extraction and Gas Chromatography
DUAN Li-cun,ZHOU Jian-yu,LI Jia-xin,LI Peng-fei,XU Fang,WANG Qi*
(School of Public Health,Kunming Medical University,Kunming 650500,China)
A method was developed for the determination of eight kinds of organochlorine pesticides,including DDTs and BHCs etc. in serum by solid-phase extraction(SPE) and gas chromatography(GC).The samples were treated by ultrasonic precipitation,cleaned on a Cleanert ODS C18N solid phase extraction column with hexane and acetone(9∶1) and purified on a Florisil solid phase extraction column.Then,the samples were blew with nitrogen until the test tubes were dry,and resolved with N-hexane.The targeted compounds were analyzed by GC with electron capture detector(ECD) .The calibration curves were linear in the range of 2-200 ng/mL,with correlation coefficients(r) of 0.996 4-0.999 0.The limits of detection(LOD) were in the range of 0.1-0.9 ng/mL,and the limits of quantitation(LOQ) were in the range of 0.4-3.0 ng/mL.The results showed that the spiked recoveries were in the range of 80.5%-112.7% with relative standard deviations(RSD)of 2.1%-7.9%.The method has high accuracy and precision,and could be used for the analysis of trace organochlorine pesticides in serum samples.
serum;DDTs;BHCs;organochlorine pesticides;solid-phase extraction(SPE);gas chromatography(GC)
2016-04-12;
2016-06-28
10.3969/j.issn.1004-4957.2016.12.016
O657.71;F767.2
A
1004-4957(2016)12-1606-05
*通讯作者:王 琦,博士,副教授,研究方向:营养与食品卫生学,Tel:0871-65922924,E-mail:lwangqi@163.com
