应用ABEEMσπ极化力场对Zn2+水溶液配位微结构和水交换反应进行分子动力学模拟研究
2016-12-29赫兰兰姜新蕊杨忠志赵东霞
赫兰兰 郭 宇 赵 健 姜新蕊 杨忠志 赵东霞
(辽宁师范大学化学化工学院,辽宁大连116029)
应用ABEEMσπ极化力场对Zn2+水溶液配位微结构和水交换反应进行分子动力学模拟研究
赫兰兰 郭 宇 赵 健 姜新蕊 杨忠志*赵东霞*
(辽宁师范大学化学化工学院,辽宁大连116029)
应用ABEEMσπ极化力场,对Zn2+水溶液体系进行分子动力学模拟,探讨Zn2+的配位微结构和配体水交换反应。水分子模型采用ABEEM-7P精细水模型。模拟后对体系结构、电荷及动力学性质进行细致分析。结构分析表明,平衡体系中Zn2+的第一层配位数为6,这与实验值是一致的。水交换反应过程中,溶剂水由O―Zn―O角分线斜上(下)方进攻Zn2+,配位水由O―Zn―O角分线斜下(上)方逐渐远离。极化力场模拟时Zn2+与交换水间的距离变化波动较大,而固定电荷力场的波动较小。模拟发现,极化力场的径向分布函数能精细地展示第二、三层配体的配位微结构,第二配位层存在靠近Zn2+的亚壳层,能与第一水合层发生水交换反应,充分体现了Zn2+的极化效应。本文阐明了水交换反应中,Zn2+位点电荷与交换水中氧原子孤对电子位点电荷的规律性变化,从电荷的角度解释了水交换反应的合理性。ABEEMσπ极化力场模拟Zn2+水溶液获得第一水合层的平均配位驻留时间为2.0×10-9s,在实验值范围内,说明ABEEMσπ极化力场可以合理地模拟Zn2+水溶液体系。
ABEEMσπ极化力场;分子动力学模拟;水交换反应;径向分布函数;电荷分析;平均配位驻留时间
1 引言
很多二价金属离子在蛋白中都发挥着重要的作用1-4,例如镁离子、钙离子和锌离子等。Zn2+涵盖在六大类酶中,在300多种酶中充当协调因子,同时Zn2+在体内反应中作为Lewis酸,起到催化作用。然而,Zn2+参与的这些重要的生命过程都离不开水,少部分含锌金属酶的催化过程要利用Zn2+的水交换反应来完成5,6。例如碳酸酐酶II7调节人体内的酸碱平衡就涉及到水交换过程。所以深入理解Zn2+水溶液体系的结构和动态变化至关重要。
在大多数锌的复合物中,锌的配位形式主要有四面体构型的4配位形式和八面体构型的6配位形式8。在水溶液中,Zn2+与水分子形成八面体构型。关于Zn2+水溶液体系的性质,近些年来很多实验组和理论组对其进行了相关研究5,9-16。实验上的研究方法主要有XRD(X-ray diffration),X射线吸收光谱和EXAFS(extended X-ray absorption final Structure),而理论上主要采用QM/MM-MD(quantum mechanical/molecular mechanical-molecular dynamics)和分子力场方法。对于Zn2+水溶液第一水合层配位数和Zn―O平衡距离的问题,实验和理论上都有明确的报道。例如Kuzmin等10通过EXAFS实验测得Zn2+水溶液体系中锌的第一水合层配位数为6.0±0.2,Zn―O平衡距离为(0.206±0.002) nm;Wu等11采用AMOEBA极化力场模拟出Zn2+第一水合层有6个水,同时在径向分布函数(RDF)分析中Zn―O的第一峰值出现在0.198 nm处;Fatmi等15,17采用QM/MM-MD方法也模拟出Zn2+的第一水合层配位数是6,在RDF中Zn―O的平衡距离为0.211nm;Mohammed等16分别应用QM/MM-MD和经典动力学模拟(CMD)方法同样也模拟出第一水合层有6个水,他们模拟出Zn―O的RDF第一峰值分别出现在0.216和0.223 nm。从上面研究可以发现Zn2+水溶液中第一水合层配位数为6,Zn―O平衡距离在0.198-0.223 nm之间。对于第二水合层水分子数,实验和理论上没有统一的标准。XRD实验测出了Zn2+的第二层配位数范围为8-13,并且第二层配体氧距Zn2+的距离为0.421-0.426 nm;Fatmi等15认为第二层配位数范围为10-18,大多数情况是14,RDF显示第二水合层的峰值为0.440 nm;Mohammed等16采用QM/MM-MD和CMD的方法模拟出第二层配体数分别为13和16,RDF的第二峰值分别出现在0.454和0.469 nm;Marini等18通过蒙特卡洛模拟得到第二水合层水分子数在20-22之间,水中氧原子距Zn2+的距离在0.430-0.440 nm之间。除此之外,对Zn2+水溶液的光谱信息、水分子的驻留时间和速率常数等也有报道。
然而,离子水溶液体系存在着一些极其有趣的动态变化,例如水交换反应,这一过程实验上是很难捕获到的。水交换反应存在着以下几种机制,分别是:缔合交换(A机制)、解离交换(D机制)、相互交换(I机制)、Ia机制和Id机制。该性质可以通过动力学模拟来实现。水交换过程必然会引起交换水分子的电荷重新分布,产生相应的极化效应,这一点是固定电荷力场不能描述的,可极化力场可以洞悉。目前,模拟水溶液体系的极化力场主要是AMOEBA极化力场11,12,19,20,该力场中考虑了二体相互作用和三体相互作用,虽然可以较精确地模拟离子水溶液体系,但是计算多级相互作用要消耗大量的时间,而且还不能应用到生物大分子体系的研究。Yang等21-25建立和发展的ABEEMσπ极化力场,将体系的电荷划分到原子、键以及孤对电子位点上,可以真实地反映出分子中各位点电荷随环境的改变,通过电荷分布的变化和转移反映出极化效应的强弱。目前该力场不仅可以快速计算有机和生物大分子体系的总能量和电荷分布等性质,也成功地应用于水分子团簇26和纯水溶液、离子水溶液体系27-30、多肽和蛋白质31,32以及核酸等体系。
本文应用ABEEMσπ极化力场对Zn2+水溶液的配位微结构和水交换反应进行研究,水溶液的水交换反应见式(1),其中水分子采用ABEEM-7P模型,即将水分子划分成七个位点,分别是三个原子位点,氧原子的两个孤对电子位点以及两个O―H间键位点。讨论了极化力场模拟平衡后,Zn2+溶液体系的水交换反应过程中显性水电荷的变化规律,Zn2+各配位层水分子的微结构,第一水合层水分子的平均配位驻留时间以及水交换反应的速率常数。

2 研究方法
2.1 ABEEMσπ极化力场
Zn2+水溶液体系的ABEEMσπ总能量,EABEEMσπ表达式为:

上式中Eb是键长伸缩振动能,Zn2+与配位水分子中氧原子之间采用Morse势能函数,溶剂水分子的键长伸缩振动采用Morse势能函数;公式(3)为该体系键长伸缩振动能的表达式。

Eθ是键角弯曲振动能,表达式如公式(4)

公式(3-4)中r是实际的键长,req是平衡的键长,θ是实际的键角,θeq是平衡的键角,kθ表示力场的键角力常数;D是键的解离能,α是和键长力常数相关的参数(α=(kb/2D)1/2,kb为键长伸缩振动力常数)。
EvdW和Eelec是非键相互作用势能。其中vdW相互作用EvdW的表达式为:

公式(5)中,εab、σab和rab分别表示a与b间的势阱深度、碰撞直径和距离。此处采用了标准的联合规则:εab=(εaaεbb)1/2,σab=1/2(σab+σab),fab表示常数。在分子中,处在不同位置的a-b原子间的fab是不同的,1-2关系和1-3关系的fab=0.0,1-4关系的fab= 0.5,其他情况下fab=1.0。
ABEEMσπ模型的独特之处在于静电相互作用项Eelec

公式(6)中,qi和qj分别是位点i和j的电荷,rij是位点i与j间的距离。当i和j之间的最短连接路径关系(包括σ键位点)小于1-6关系时,kij=0;当i和j在氢键相互作用区域时,kij=kH-bond22,23;其它所有情况,kij=0.57。
2.2 ABEEMσπ极化力场参数和ABEEM-7P水模型
ABEEMσπ极化力场中,分子的电荷位点包括原子位点、键(σ/π键)位点和孤对电子位点,各原子周围键和孤对电子位点的电荷一般是各向异性的,不对称的。各位点的电荷是应用电负性均衡方法,求解电负性均衡方程得到的,具体的能量公式和方程表达式请见文献21-23。
ABEEMσπ极化分子力场拟合出了一套适用于Zn2+水溶液体系的参数。表1列出了Zn2+水溶液体系的电荷参数(价态电负性χ*和价态硬度2η*)、键长参数(平衡键长req,键的解离能D,以及与键长力常数相关的参数α)、键角参数(平衡键角θeq和键角力常数kθ)以及范德华参数(碰撞直径σ和势阱深度ε)。溶剂水的电荷、范德华、键长和键角参数仍采用原来ABEEMσπ力场的参数23,33,其余参数是最新拟合得到的。本文中考虑到第一层配体水分子与溶剂水分子所处的化学环境不同,受到锌离子的静电极化作用较强,为了更接近真实情况,第一层配体水分子和溶剂水分子采用不同的参数。当配体水分子和溶剂水分子发生交换时,水分子参数也发生相应的变化。
为了使这套参数不仅适用于Zn2+水溶液体系,而且也适用于我们将来要研究的含Zn2+蛋白体系。在确定电荷参数时,我们以B3LYP/6-311++G(d,p)水平下优化后的以及PDB数据库中截取的三十多种含Zn2+体系作为模型分子,包含二、三、四、五、六个配位数,配体主要为氨基酸侧链上的原子、水分子和抑制剂分子,Zn2+的基点电荷是1.0。采用从头算的方法获得模型分子中各原子的Mulliken电荷,并以此为标准,利用组内编写的程序,根据线性回归和最小二乘法拟合出了一套适用于Zn2+水溶液体系以及绝大多数含锌蛋白电荷分布计算的参数。
Morse势能参数的拟合,选取B3LYP/6-311++ G(d,p)水平下优化后的Zn2+-His-2Cys-H2O为模型分子,并利用从头算的方法拟合Zn2+与水分子间的Morse势能曲线,获得Zn―O的Morse势能参数;将参数加入ABEEMσπ力场中,优化1-6)的稳定结构和PDB数据库中截取的Zn2+配体中含水分子的结构。反复调节参数,获得优化后使体系Zn―O键长变动最小的参数。配体水的O―H键解离能仍采用溶剂水的O―H键解离能,平衡键长做了微调,获得了适用于第一层配体水分子的平衡键长。
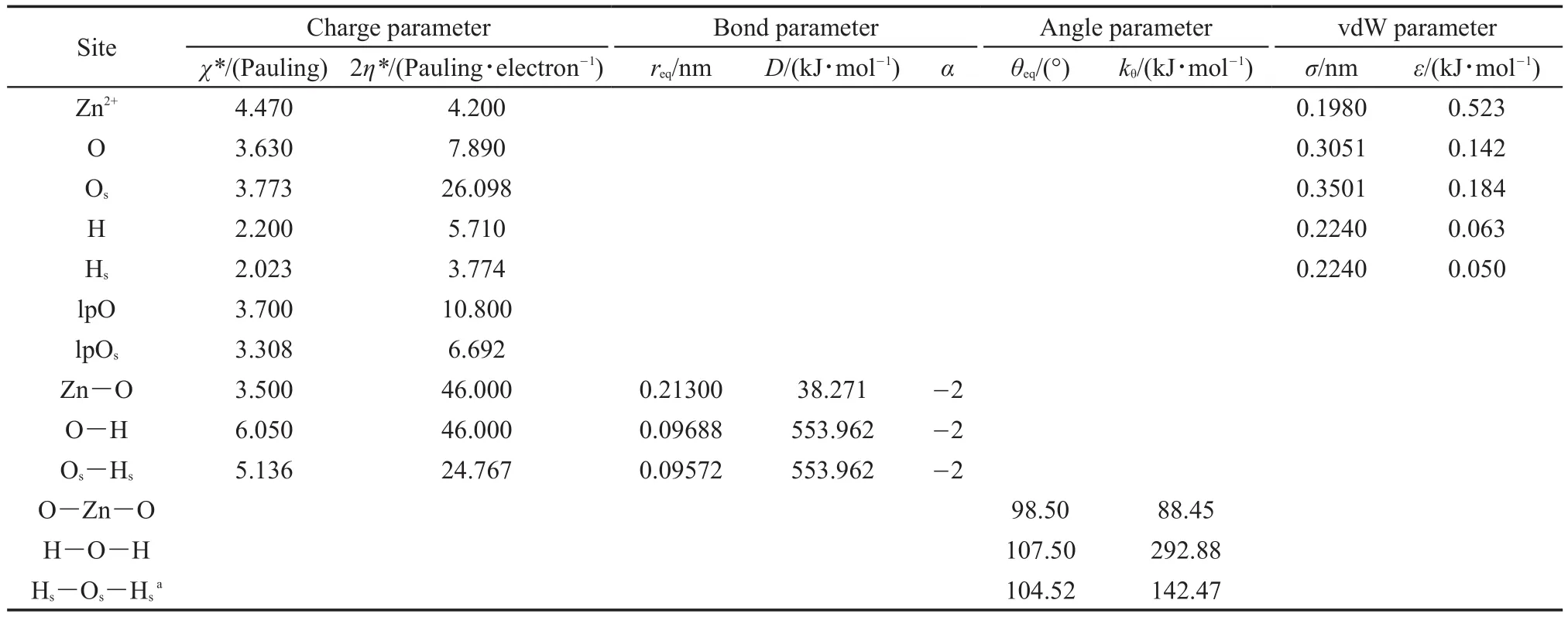
表1 Zn2+水溶液体系ABEEMσπ力场参数Table 1 Parameters of Zn2+aqueous solution forABEEMσπ force field
键角参数的拟合,选取Zn2+-His-2Cys-2(H2O)为模型分子,采用上述方法进行优化和频率计算,获得稳定的结构。分别对∠O―Zn―O和∠H―O―H每偏离平衡位置1°保存一个结构,共保存20个结构,拟合出∠O―Zn―O和∠H―O―H的势能曲线,并获得平衡键角和键角力常数。将参数加入ABEEMσπ力场中,以从头算获得的水合锌离子以及PDB中截取的含Zn2+体系为标准,采用ABEEMσπ力场优化上述结构,对比优化前后的结构,反复调节参数,获得优化后使∠O―Zn―O和∠H―O―H变动最小的参数。
Li等34报道的Zn2+的vdW参数表明,Zn2+的vdW参数受到金属离子在力场中的模型、水分子模型和模拟环境等因素的影响,Zn2+的vdW参数可转移性会受到局限。本文参考了Santosh等35在AMBER力场中进行分子动力学模拟时,研究二肽和Zn2+水溶液体系所用的Zn2+的vdW参数,碰撞直径σ为0.20471 nm,势阱深度ε为0.569 kJ·mol-1。以动力学模型平衡时Zn2+的配位结构与实验数据相近为标准,在Santosh等人的Zn2+的vdW参数附近调节,获得适用于ABEEMσπ极化力场的Zn2+的vdW参数。配体水中O和H的范德华参数是在溶剂水中O和H的范德华参数的基础上微调得到的。
水分子七点极化力场非刚体模型(ABEEM-7P)22,23示于图1。水分子的七点模型包含三个原子位点,两个O―H化学键位点和两个孤对电子位点。单体水中O―H键长和H―O―H键角分别设为相应的实验值0.09572 nm和104.5°,O―H键位点设在氢原子和氧原子的共价半径之比处,孤对电子的位点在距氧原子0.074 nm处。水分子中正电荷集中在氧原子和氢原子上,而负电荷分布在孤对电子区域和化学键区域。ABEEM-7P水模型不同于前人的TIP4P、POL5和其他的水分子模型,它具有两个显著的特点:(1)ABEEM-7P水模型提供了更大的适用范围,它不仅可以计算原子、化学键和孤对电子的电荷,也提出了氢键相互作用区域的概念,通过引入氢键拟合函数kH-bond来描述所形成氢键的孤对电子和水中氢原子间的静电相互作用,使氢键信息描述得更加精确;(2)在分子力学中,水分子为非刚体结构,允许键长和键角发生振动,键长伸缩振动使用Morse势能函数来描述,因为它能够从平衡键长到键的解离更大范围描述键的伸缩振动。范德华相互作用采用12-6的Lennard-Jones势能函数。ABEEM-7P模型可以很清晰地描述电荷随环境的变化和氢键信息,所以使用该模型讨论Zn2+水溶液的结构和性质。
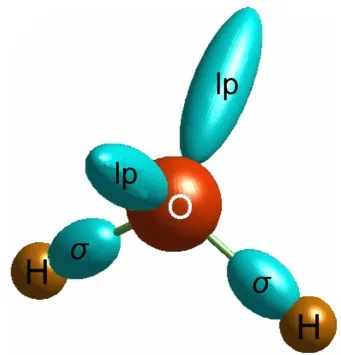
图1水分子的ABEEM-7P模型示意图Fig.1 Diagram ofABEEM-7Pwater molecular model
2.3 平均配位驻留时间
水分子在离子周围的平均配位驻留时间36可以通过时间相关函数来得到:
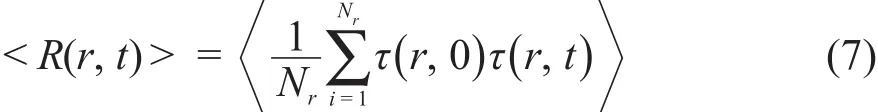
公式(7)中τ(r,t)是Heaviside单位函数。如果一个水分子i在半径为r的区域范围内,τ(r,t)等于1;如果一个水分子i不在半径为r的区域范围内,τ(r,t)等于0。本文中,计算Zn2+第一层配体水的平均配位驻留时间时,r=0.300 nm;第二层时,r在0.345-0.560 nm范围内;第三层时,r在0.560-0.800 nm范围内。Nr代表t=0时被统计区域内水分子的个数,同时允许水分子短暂离开区域r的最长时间不超过2 ps。如果水分子在离子周围的停留时间较长,2 ps的时间不会影响统计计算结果。
2.4 过渡态理论速率常数
过渡态理论的速率常数20表达式为:

其中300 K时校正因子k0≈(kBT/h)≈0.6×1013s-1,R=8.314×10-3kJ·mol-1·K-1,ΔG≠是发生水交换反应的活化能,通过分析平均力势(PMF)36获得,平均力势及有效平均力势(EPMF)可以通过径向分布函数获得,如公式(9-10):
其中β=1/(kBT),kB为玻尔兹曼因子,T为热力学温度,r代表离子与水分子中氧原子间的距离,r*代表活化势垒的位置,W(r)和Weff(r)分别表示平均力势和有效平均力势。
2.5 模拟细节
将Zn2+融入Tinker程序中具有500个水分子的waterbox立方体水盒子中,模拟体系中水分子的个数为497个,加入两个抗衡离子Cl-,保持体系净电荷为0。体系盒长为2.4662 nm。ABEEMσπ力场中各区域划分情况见图2,包括原子、键(Zn―O/O―H键)和孤对电子区域。分别采用ABEEMσπ固定电荷和极化力场的成键模型,对建好的模拟体系进行优化和动力学模拟。采用正则系综(NVT),利用Berendsen热浴控制每个分子的温度,并利用Verlet跳蛙法积分运动方程,选取积分步长为1 fs,极化力场每隔1 ps重新计算体系中各位点的电荷,各体系每隔1 ps保存一次坐标文件,模拟时间为2 ns。考虑周期性边界条件和最近镜像,采用截断半径方法(截断半径均为1.2331 nm),并且利用开关函数调节非键相互作用。在整个模拟过程中,对Zn2+和抗衡离子进行固定,其余原子可以自由移动。模拟初速度通过Maxwell-Boltzmann分布随机给出,模拟过程中原子的运动遵守牛顿运动方程,通过调节速度使体系达到设定的温度300 K,整个体系保持局域守恒。

图2 ABEEMσπ力场中Zn(H2O)62+各区域示意图Fig.2 Diagram of Zn(H2O)62+divided into regions in ABEEMσπ force field
3 结果与讨论
3.1 结构性质及电荷分析
3.1.1 捕获水交换反应

图3 ABEEMσπ力场模拟Zn2+水溶液体系过程直接观测到水交换反应Fig.3 Direct observation of the water exchange events near Zn2+in water based onABEEMσπ force field
3.1.2 ABEEMσπ极化力场水交换反应过程中电荷的变化
在水交换反应的过程中,交换水中氧原子孤对电子位点电荷也发生了规律性的变化,下面具体讨论了水溶液体系水交换反应过程中电荷的变化。

表2 ABEEMσπ极化力场动力学模拟时间在1-15 ps内,Zn(H2O)62+水溶液体系水中氧原子与Zn2+间的距离变化情况Table 2 Distance between Zn2+and oxygen atom of water,during dynamic simulation time 1-15 ps, based onABEEMσπ polarizable force field

图4 ABEEMσπ极化力场Zn2+第一层配体水交换的结构示意图Fig.4 Structure diagrams of the first shell ligand exchange events near Zn2+in water based on ABEEMσπ polarizable force field

表3 ABEEMσπ极化力场动力学模拟时间为1-15 ps内,Zn(H2O)26+水溶液体系中Zn2+位点电荷及交换水中氧原子的孤对电子位点电荷Table 3 Charge of Zn2+site and lone pair site of oxygen atom of exchange water,during dynamic simulation time 1-15 ps,based onABEEMσπ polarizable force field
模拟时间为2-3 ps的过程中发生了第二次水交换反应(图4(c)是模拟时间为3 ps时的结构)。O421处于Zn2+的一、二配位层之间,从O496―Zn―O1177角分线的斜下方进攻Zn2+,到达了Zn2+的第一配位层,占据O1177的位置,O421孤对电子位点电荷获得了-0.059e。同时O1177从右下方逐渐远离Zn2+,到达了Zn2+的第二配位层,距Zn2+0.411 nm,O1177孤对电子位点电荷失去了-0.078e。
模拟时间为5-6 ps过程中发生了第三次水交换反应。图4(d)和(e)分别代表模拟时间为5和6 ps时的结构。5 ps时,O496、O343和O421与Zn2+距离分别为0.270、0.245、0.261 nm,此时形成七配位结构,Zn2+的杂化方式为sp3d3杂化,但这个结构并没有稳定存在。模拟时间为6 ps时,O496与Zn2+间的距离为0.446 nm;而O343和O421仍为Zn2+的第一层配体,与Zn2+距离分别为0.213、0.212 nm;此时Zn2+的杂化方式为sp3d2。在这个水交换反应过程中交换水的孤对电子位点电荷发生了规律性的变化。模拟时间为4-6 ps,O496由Zn2+的第一配位层运动到第二配位层,这个过程中O496的孤对电子位点电荷由-0.515e变为-0.455e。5-6 ps时,O421和O343孤对电子位点电荷分别增加了-0.036e和-0.049e,同时Zn2+上的电荷也由1.036e变为1.066e,增加了0.03e。这是由于Zn2+与交换水距离变短,它们之间的静电吸引作用加强。
模拟时间为11-12 ps时,发生了第四次水交换反应。仍可得到和上述一致的结论,从电荷的角度分析,处于Zn2+第二配位层之内的水分子受到Zn2+的静电极化作用较强;靠近Zn2+时,交换水中O的孤对电子位点负电荷电量增大;远离Zn2+时,交换水中O的孤对电子位点负电量减少,这样更容易发生水交换反应。
ABEEMσπ极化力场模拟Zn2+水溶液体系时,在水交换反应过程中Zn2+与交换水孤对电子位点电荷发生了规律性的变化,从电荷的角度解释了水交换反应的发生,这是固定电荷力场和AMOEBA极化力场不能实现的。
3.1.3 径向分布函数
为了描述ABEEMσπ力场中Zn2+周围水分子的微观结构,我们分析了动力学模拟平衡后Zn―O之间的径向分布函数(RDF)。图5中黑线和红线分别表示ABEEMσπ固定电荷和极化力场模拟Zn2+水溶液体系的Zn―O径向分布函数图。表437中列出了ABEEMσπ固定电荷及极化力场模拟Zn2+水溶液体系平衡后,Zn2+各配位层的配位数及RDF的峰值位置。从图5中可以看出固定电荷力场与极化力场获得的RDF有很大区别。极化力场RDF的第一峰要比固定电荷力场的高出一倍,说明极化力场Zn2+与第一层配体之间的相互作用强于固定电荷力场的。固定电荷和极化力场获得的RDF第一峰的积分数都是6,说明Zn2+在水溶液中以六配位的形式存在较为稳定,这与实验值10是一致的。ABEEMσπ极化力场的RDF的第二层配体的分布情况不同于ABEEMσπ固定电荷力场和AMOEBA极化力场11;ABEEMσπ固定电荷力场以及AMOEBA极化力场的RDF第二峰是一个较为平缓的峰,而ABEEMσπ极化力场第二层配体存在几个亚壳层,并且是很陡的尖峰,充分展现了距Zn2+一定距离的很小壳层内水分子的分布有一定秩序;这种微结构体现了Zn2+对水溶液体系的极化效应。ABEEMσπ固定电荷力场第二层配体氧与Zn2+距离的范围为0.320-0.510 nm,第二层配位数约为18.2个;极化力场第二层配体与Zn2+间距离的范围为0.348-0.560 nm,共28个水分子,与Zn2+距离范围为0.345-0.380 nm的亚壳层内有7个水分子,这些水分子可与第一层配体发生水交换反应。相比于ABEEMσπ固定电荷力场,ABEEMσπ极化力场配体与Zn2+距离0.628 nm处出现了第三峰,第三层配体的配位数约为50.0个。以上分析表明ABEEMσπ极化力场模拟Zn2+溶液与固定电荷力场以及AMOEBA极化力场相比更精确。
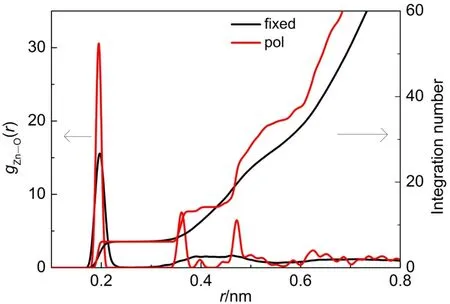
图5 ABEEMσπ力场Zn2+水溶液体系Zn―O径向分布函数(RDF)以及积分线Fig.5 RDFs of Zn―O and integration lines for Zn2+aqueous solution based onABEEMσπ force field color online
3.1.4 角度分布函数
图6是ABEEMσπ固定电荷力场和极化力场模拟Zn2+水溶液体系平衡后O―Zn―O的角度分布函数图。从图中可以看出固定电荷力场和极化力场模拟Zn2+水溶液体系平衡后,O―Zn―O的角度分布函数图中有两个明显的峰,固定电荷力场得到的两个峰值分别出现在88.5°和171.5°,峰的范围较宽且存在小的肩峰,是较为扭曲的八面体构型。而极化力场得到的两个峰分别出现在89.5°和178.5°,并且峰的宽度较小,是比较规则的八面体构型。另外,极化力场获得的O―Zn―O角度分布函数的峰值远大于固定电荷力场的峰值。说明极化力场所模拟的Zn2+水溶液体系形成6配位的八面体构型的几率更大。

表4 ABEEMσπ固定电荷力场和极化力场模拟Zn2+水溶液体系平衡后,Zn2+各配位层的配位数及Zn―O的RDF出峰位置与AMOEBA、QM/MM和EXAFS对比Table 4 Coordination number of different hydration shells and the peaks of Zn―O RDFs in Zn2+aqueous solution equilibrium system based onABEEMσπ fixed force field and polarizable force field,AMOEBA,QM/MM and EXAFS
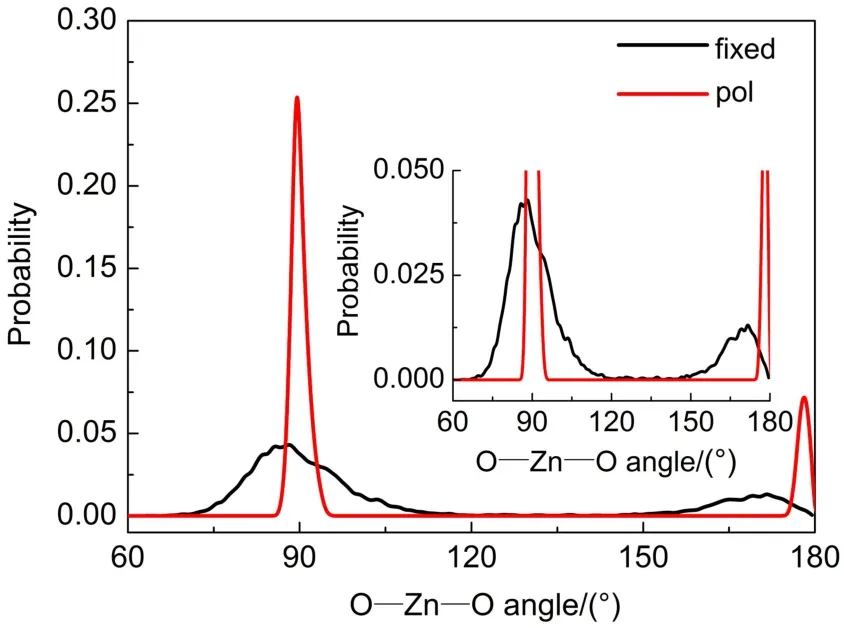
图6 ABEEMσπ力场Zn2+水溶液体系O―Zn―O角度分布函数图Fig.6 Angle distribution function of O―Zn―O in Zn2+aqueous solution based onABEEMσπ force field
3.2 动力学性质分析
3.2.1 平均配位驻留时间的统计
通过平均配位驻留时间的相关函数可获得Zn2+第一水合层水分子的驻留时间(见公式(7))。经统计ABEEMσπ可极化力场中Zn2+水溶液的第一层配体水分子的平均配位驻留时间为2.0×10-9s,实验值38在10-10-10-9s之间。AMOEBA极化力场11Zn2+溶液第一层配体水的平均驻留时间为2.2× 10-9s,Classical固定电荷力场11分子动力学模拟得到第一层配体的平均驻留时间约为1.5×10-10s。ABEEMσπ可极化力场获得的Zn2+第一水合层的平均配位驻留时间与其他力场相差不大,且在实验值范围内。ABEEMσπ可极化力场模拟Zn2+水溶液体系得到第二、三层配体水分子的平均配位驻留时间分别为4.3×10-10和4.0×10-10s。从上述数据可以看出,ABEEMσπ极化力场外层水的平均配位驻留时间要比内层水的短,说明外层水分子受到Zn2+的极化作用小。
3.2.2 水交换反应速率常数的统计

图7 ABEEMσπ极化力场Zn2+与水分子的平均力势(W(r))和有效平均力势(Weff(r))随Zn2+和水分子间的距离变化情况Fig.7 Potential of mean force(W(r))and effective potential of mean force(Weff(r))between Zn2+and water with the distance change between Zn2+and water based on ABEEMσπ polarizable force field
图7展示了Zn2+与水分子间的平均力势和有效平均力势随Zn2+与水分子间的距离变化情况。从图中可以获悉ABEEMσπ极化力场模拟Zn2+水溶液体系,获得Zn2+与第一水合层水分子的解离能垒约为7.8kBT(1kBT约为2.5 kJ·mol-1)。与其他固定电荷力场相比,ABEEMσπ极化力场的优势是不仅可以获得第一层配体水交换反应的活化能垒,而且可以获得第二、三层配体水交换的活化能垒,它们分别约为0.5kBT和1.0kBT。ABEEMσπ极化力场的PMF活化能垒中,第一水合层的水分子与Zn2+之间的解离能垒最大,这是由于第一水合层的水分子与Zn2+间的相互作用较强;第二层配体水分子发生水交换反应的能垒最小,说明第二层配体很容易与第一层配体和第三层配体发生水交换反应,自由移动性强;第三层配体水交换反应能垒比第二层配体的大,这是由于第三层配体间水分子的平均氢键数目比第二层配体的多,水分子移动时要克服的氢键相互作用较大。ABEEMσπ极化力场可以精细地反映各层配体水发生水交换反应的难易情况,与其特有的极化力场和ABEEM-7P水模型密切相关。
由平均力势分析获得ABEEMσπ极化力场第一水合层水交换反应的活化能为19.5 kJ·mol-1,Michael等39采用从头算的方法获得气相下Zn2+第一水合层水交换反应的活化能垒在17.6-19.2 kJ·mol-1之间,ABEEMσπ极化电荷力场获得的第一水合层水交换反应的活化能与从头算结果非常接近,说明ABEEMσπ极化电荷力场模拟Zn2+水溶液体系的结果是合理的。通过公式(8)获得极化力场过渡态理论的速率常数为2.4×109s-1;其倒数与我们获得的第一层配体水分子的平均配位驻留时间吻合。实验报道40,298 K时Zn2+水溶液中第一层配体水交换反应的速率常数约为2×107s-1。温度升高水交换反应的速率会加快,这说明ABEEMσπ力场获得300 K时,Zn2+第一水合层水交换反应的速率常数的数量级是合理的。
4 结论
应用ABEEMσπ固定电荷力场和极化力场模拟Zn2+水溶液体系,获得的Zn2+第一水合层的配位数与实验值一致。对RDF进行分析,独特的ABEEMσπ极化力场和ABEEM-7P精细水模型模拟Zn2+水溶液发现了第一、二和三层配体水分子的微结构,Zn2+的第二层配体存在亚壳层,可与第一层配体发生水交换反应。ABEEMσπ极化力场模拟Zn2+溶液体系获得了水交换反应过程中Zn2+位点与交换水分子孤对电子位点电荷的规律性变化,解释了水交换反应的合理性。ABEEMσπ极化力场Zn2+水溶液体系第一水合层水分子平均配位驻留时间在实验值范围内,并且第一水合层水分子的解离能垒与从头算很接近,说明了ABEEMσπ极化力场模拟Zn2+水溶液体系的结果是合理的。本文对Zn2+水溶液体系的配位微结构以及水交换反应的探讨,为进一步研究其它过渡金属离子水溶液中的水交换反应,以及一些酶催化过程中的水交换反应奠定基础。
致谢:我们对Ponder教授提供的Tinker源程序表示衷心的感谢。
(1) Cowan,J.A.Chem.Rev.1998,98(3),1067.doi:10.1021/ cr960436q
(2) Silverman,D.N.;McKenna,R.Accounts Chem.Res.2007,40 (8),669.doi:10.1021/ar7000588
(3) Imanishi,M.;Matsumura,K.;Tsuji,S.;Nakaya,T.;Negi,S.; Futaki,S.;Sugiura,Y.Biochemistry 2012,51(16),3342. doi:10.1021/bi300236m
(4) Kepp,K.P.Chem.Rev.2012,112(10),5193.doi:10.1021/ cr300009x
(5) Ohtaki,H.;Radnai,T.Chem.Rev.1993,93(3),1157. doi:10.1021/cr00019a014
(6) Helm,L.;Merbach,A.E.Coord.Chem.Rev.1999,187(1), 151.doi:10.1016/s0010-8545(99)90232-1
(7) Nielsen,P.M.;Fago,A.J.Inorg.Biochem.2015,149,6. doi:10.1016/j.jinorgbio.2015.04.013
(8) Maynard,A.T.;Covell,D.G.J.Am.Chem.Soc.2001,123(6), 1047.doi:10.1021/ja0011616
(9) Asthagiri,D.;Pratt,L.R.;Paulaitis,M.E.;Rempe,S.B.J.Am. Chem.Soc.2004,126,1285.doi:10.1021/ja0382967
(10) Kuzmin,A.;Obst,S.;Purans,J.J.Phys.:Condens.Matter 1997,9,10065.
(11) Wu,J.C.;Piquemal,J.P.;Chaudret,R.;Reinhardt,P.;Ren,P.Y. J.Chem.Theory Comput.2010,6(7),2059.doi:10.1021/ ct100091j
(12) Xiang,J.Y.;Ponder,J.W.J.Comput.Chem.2013,34(9),739. doi:10.1002/jcc.23190
(13) Babu,C.S.;Lim,C.J.Phys.Chem.A 2006,110(2),691. doi:10.1021/jp054177x
(14) Fatmi,M.Q.;Hofer,T.S.;Randolf,B.R.;Rode,B.M.J.Phys. Chem.B 2008,112(18),5788.doi:10.1021/jp710270z
(15) Fatmi,M.Q.;Hofer,T.S.;Randolf,B.R.;Rode,B.M.J.Phys. Chem.B 2006,110(1),616.doi:10.1021/jp0546655
(16) Mohammed,A.M.;Loeffler,H.H.;Inada,Y.;Tanada,K.; Funahashi,S.J.Mol.Liq.2005,119(1-3),55.doi:10.1016/j. molliq.2004.10.008
(17) Fatmi,M.Q.;Hofer,T.S.;Randolf,B.R.;Rode,B.M.J.Phys. Chem.B 2007,111(1),151.doi:10.1021/jp0654213
(18) Marini,G.W.;Texler,N.R.;Rode,B.M.J.Phys.Chem.1996, 100(16),6808.doi:10.1021/jp953375t
(19) Soniat,M.;Hartman,L.;Rick,S.W.J.Chem.Theory Comput. 2015,11(4),1658.doi:10.1021/ct501173n
(20) Kurnikov,I.V.;Kurnikova,M.J.Phys.Chem.B 2015,119(32), 10275.doi:10.1021/acs.jpcb.5b01295
(21) Zhao,D.X.;Liu,C.;Wang,F.F.;Yu,C.Y.;Gong,L.D.;Liu,S. B.;Yang,Z.Z.J.Chem.Theory Comput.2010,6(3),795. doi:10.1021/ct9006647
(22) Yang,Z.Z.;Wu,Y.;Zhao,D.X.J.Chem.Phys.2004,120(6), 2541.doi:10.1063/1.1640345
(23) Wu,Y.;Yang,Z.Z.J.Phys.Chem.A 2004,108(37),7563. doi:10.1021/jp0493881
(24) Li,X.;Yang,Z.Z.J.Phys.Chem.A 2005,109(18),4102. doi:10.1021/jp0458093
(25) Yang,Z.Z.;Cui,B.Q.J.Chem.Theory Comput.2007,3(4), 1561.doi:10.1021/ct600379n
(26) Qian,P.;Yang,Z.Z.Acta Phys.-Chim.Sin.2006,22(5),561. [钱 萍,杨忠志.物理化学学报,2006,22(5),561.] doi:10.3866/PKU.WHXB20060510
(27) Gong,L.D.Sci.Sin.Chim.2013,43(5),519.[宫利东.中国科学:化学,2013,43(5),519.]doi:10.1007/s11426-012-4787-3
(28) Li,X.;Yang,Z.Z.J.Chem.Phys.2005,122(8),084514. doi:10.1063/1.1853372
(29) Guo,C.;Liu,C.;Yang,Z.Z.Acta Phys.-Chim.Sin.2010,26(2),478.[郭 慈,刘 翠,杨忠志.物理化学学报,2010,26 (2),478.]doi:10.3866/PKU.WHXB20100219
(30) Liu,Y.;Wang,F.F.;Yu,C.Y.;Liu,C.;Gong,L.D.;Yang,Z.Z. Acta Phys.-Chim.Sin.2011,27(2),379.[刘 燕,王芳芳,于春阳,刘 翠,宫利东,杨忠志.物理化学学报,2011,27(2), 379.]doi:10.3866/PKU.WHXB20110233
(31) Zhang,Q.;Yang,Z.Z.Chem.J.Chin.Univ.2005,26(12), 2345.[张 强,杨忠志.高等学校化学学报,2005,26(12), 2345.]doi:10.3321/j.issn:0251-0790.2005.12.029
(32) Guan,Q.M.;Cui,B.Q.;Zhao,D.X.;Gong,L.D.;Yang,Z.Z. Chin.Sci.Bull.2008,53(8),1171.doi:10.1007/s11434-007-0493-5
(33) Yang,Z.Z.;Wu,Y.;Zhao,D.X.J.Chem.Phys.2004,120(6), 2541.doi:10.1063/1.1640345
(34) Li,P.F.;Roberts,B.P.;Chakravorty,D.K.;Merz,K.M.,Jr.J. Chem.Theory Comput.2013,9(6),2733.doi:10.1021/ ct400751u
(35) Santosh,M.S.;Lyubartsev,A.P.;Mirzoev,A.A.;Bhat,D.K.J. Phys.Chem.B 2010,114(49),16632.doi:10.1021/jp108376j
(36) Spångberg,D.;Rey,R.;Hynes,J.T.;Hermansson,K.J.Phys. Chem.B 2003,107(18),4470.doi:10.1021/jp027230f
(37) Zhang,Q.;Yang,Z.Z.Chem.Phys.Lett.2005,403,242. doi:10.1016/j.cplett.2005.01.011
(38) Salmon,P.S.;Bellissent-Funel,M.C.;Herdman,G.J.J.Phys.: Condens.Matter 1990,2,4297.doi:10.1088/0953-8984/2/18/ 027
(39) Michael,H.;Clark,T.;Eldik,R.V.J.Am.Chem.Soc.1997,119 (33),7843.doi:10.1021/ja970483f
(40) Akesson,R.;Pettersson,L.G.M.;Sandstroem,M.;Siegbahn,P. E.M.;Wahlgren,U.J.Phys.Chem.1993,97(15),3765. doi:10.1021/j100117a023
Study on Coordination Microstructure and Water Exchange Reaction of Zn2+Aqueous Solutions through Molecular Dynamics Simulations Using the ABEEMσπ Polarizable Force Field
HE Lan-Lan GUO Yu ZHAO Jian JIANG Xin-Rui YANG Zhong-Zhi*ZHAO Dong-Xia*
(School of Chemistry and Chemical Engineering,Liaoning Normal University,Dalian 116029,Liaoning Province,P.R.China)
Coordination microstructure and water exchange of Zn2+aqueous solution were studied through molecular dynamics simulations based on the ABEEMσπ polarizable force field with the precise ABEEM-7P model.Structural and dynamical properties were investigated in detail.We show that the first-shell water coordination number is six,which is in agreement with the experimental value.During the water exchange process,H2O,which attacks or leaves the first solvation shell of Zn2+,is orientated on the upper or lower inclined side of the∠O―Zn―O bisector.The distance change between Zn2+and the oxygen atom of the exchange water for the polarizable force field fluctuates more than that observed for the fixed charge force field.The radial distribution function(RDF)of the polarizable force field showed clearly the microstructure of the second and third solvation shells.The subshell of the second solvation shell was found to exchange with the first solvation shell.The polarizable effect of Zn2+has been fully expressed.The charges of the Zn2+site and the lone-pair sitesin exchange water regularly change,demonstrating the rationality of water exchange.The mean ligand residence time(2.0×10-9s)of the first-shell water produced by theABEEMσπ polarizable force field is within the range of the experimental value.Zn2+aqueous solution can be reasonably simulated through theABEEMσπ polarizable force field.
ABEEMσπ polarizable force field;Molecular dynamic simulation;Water exchange reaction; Radial distribution function;Charge analysis;Mean ligand residence time
O641
10.3866/PKU.WHXB201609193
Received:August 9,2016;Revised:September 19,2016;Published online:September 19,2016.
*Corresponding authors.YANG Zhong-Zhi,Email:zzyang@lnnu.edu.cn;Tel:+86-411-82159607.
ZHAO Dong-Xia,Email:zhaodxchem@lnnu.edu.cn.
The project was supported by the National Natural Science Foundation of China(21133005,21473083),General Project of Education Department of Liaoning Province,China(L2014426),and Laboratory Opening Program of Liaoning Normal University,China(cx20160111).
国家自然科学基金(21133005,21473083),辽宁省教育厅一般项目(L2014426)和辽宁师范大学实验室开放项目(cx20160111)资助
