黄芪药渣中阿拉伯木聚糖(AX⁃Ⅰ⁃1)的提取纯化、结构分析及体外抗氧化作用
2016-12-14王桂臻秦雪梅杜冠华
刘 磊,李 科,郝 霞,王桂臻,秦雪梅,杜冠华,张 翔
(1.山西大学化学化工学院,2.中医药现代研究中心,太原030006;3.中药功效物质与新药创制山西省重点实验室,太原030006;4.中国医学科学院、中国协和医科大学药物研究所,北京100050;5.美国路易斯维尔大学,路易斯维尔40201)
黄芪药渣中阿拉伯木聚糖(AX⁃Ⅰ⁃1)的提取纯化、结构分析及体外抗氧化作用
刘磊1,2,3,李科2,3,郝霞1,2,3,王桂臻1,2,3,秦雪梅2,3,杜冠华4,张翔5
(1.山西大学化学化工学院,2.中医药现代研究中心,太原030006;3.中药功效物质与新药创制山西省重点实验室,太原030006;4.中国医学科学院、中国协和医科大学药物研究所,北京100050;5.美国路易斯维尔大学,路易斯维尔40201)
黄芪药渣经分级提取,再通过二乙氨乙基(DEAE)⁃纤维素52阴离子交换柱层析和Sephacryl S⁃400 HR凝胶柱层析分离纯化,得到4个多糖组分AX⁃Ⅰ⁃1~AX⁃Ⅰ⁃4.对多糖AX⁃Ⅰ⁃1的理化性质和结构进行研究发现,AX⁃Ⅰ⁃1含有鼠李糖、阿拉伯糖、木糖、甘露糖、葡萄糖和半乳糖(摩尔比为0.006∶14.113∶8.284∶0.116∶0.468∶1),主链由阿拉伯糖和木糖通过β⁃(1→2),(1→3)和(1→4)苷键组成,支链由(1→4)βArap,(1→3)βGalp和(1→2)βMan组成,非还原末端由αRhap,βGclp和βGalp组成.对多糖AX⁃Ⅰ⁃1~AX⁃Ⅰ⁃4的抗氧化研究结果表明,这4个组分对羟基自由基、超氧阴离子自由基和1,1⁃二苯基⁃2⁃三硝基苯肼(DPPH)自由基均有一定的清除作用;当浓度为0.1 mg/mL时,多糖AX⁃Ⅰ⁃2~AX⁃Ⅰ⁃4对超氧阴离子的清除率是阳性对照维生素C(Vc)的2倍.
黄芪药渣;阿拉伯木聚糖;分离纯化;结构分析;抗氧化
黄芪是我国重要的大宗药材之一,始载于《神农本草经》,列为上品[1].目前,黄芪在临床上被广泛应用,素有“十方八芪”之说.据统计,国内黄芪年消耗量超过100万吨,而且逐年攀升,所以产生了大量的黄芪药渣.目前的中药药渣处理方法虽然相对于早期有很大的变化,也开展了进一步利用,如用于食用菌栽培基料、制成有机肥料、畜禽饲料及造纸等,但是这些产品附加值较低,甚至会造成环境的二次污染[2].因此,开发中药药渣高附加值产品,构建科学循环利用模式,建立循环产业链,对实现生态资源的良性循环具有深远意义.
中药药渣部分多为植物细胞壁成分,研究[3]已证实多种活性多糖蕴含在植物细胞壁组分(果胶、半纤维素)中.其中,半纤维素含有活性多糖的种类最丰富,被称为潜在的“药物资源库”[4].目前,已有对植物细胞壁中半纤维素进行结构及活性研究的相关报道.如周素梅等[5,6]对麦麸中阿拉伯木聚糖的结构及活性进行了研究,发现其具有较强的抗肿瘤活性,可同时提高机体特异和非特异性免疫系统的反应.然而,该聚糖含量仅为谷物种皮的1%~2%,含量低,提取难度大.
我们[7]在前期研究中发现,黄芪药渣细胞壁半纤维素多糖的主要成分是阿拉伯糖和木糖,推测可能为阿拉伯木聚糖,并通过提取和分离纯化,对其理化性质进行了初步研究.该组分含量约占黄芪细胞壁组分的10%,较麦麸中的阿拉伯木聚糖高约1000倍,但其结构特点并不清楚.基于此,本文针对其中一种阿拉伯木聚糖AX⁃Ⅰ⁃1,研究了其单糖组成、连接位点、NMR谱学特征及体外抗氧化活性,以期为黄芪药渣中阿拉伯木聚糖的综合开发利用奠定基础.
1 实验部分
1.1 试剂与仪器
黄芪药材为山西浑源出产的半野生蒙古黄芪,经山西大学秦雪梅教授鉴定为豆科植物黄芪属蒙古黄芪[Astragalus membranaceus(Fisch.)Bge.var.mongholicus(Bge.)Hsiao].标本保存于山西大学中医药现代研究中心.
二乙氨乙基(DEAE)⁃纤维素⁃52及Sephacryl S⁃400 HR购自北京索莱宝科技有限公司;无水二甲基亚砜(纯度99.7%,带分子筛),购自百灵威化学试剂公司;氘代硼氢化钠(NaBD4)购自上海江莱生物公司;1,1⁃二苯基⁃2⁃三硝基苯肼(DPPH,纯度95%)购自Alfa Aesar公司;氘代二甲基亚砜购自Cam⁃bridge Isotope Laboratories公司;葡聚糖标准品购自中国计量科学研究院;糖醇标准品购自Sigma公司;其它试剂均为分析纯.
Thermo Finnigan气相色谱⁃质谱联用仪(美国热电公司);Bruker Tensor 27型红外光谱仪和Bruker DRX 600 MHz核磁共振波谱仪(德国布鲁克公司);普析TU⁃1810型紫外⁃可见分光光度计(中国北京普析通用仪器有限公司);依利特P230型高效液相色谱仪(中国大连依利特分析仪器有限公司);Shodex RI⁃201H型示差折光检测器(日本昭和电工科学仪器有限公司).
1.2 黄芪药渣阿拉伯木聚糖(AXs)的提取
1.2.1 黄芪药渣的制备 将半野生蒙古黄芪粉碎,过100目筛;称取粉末20 g,加入600 mL蒸馏水,于42℃水浴加热1 h;离心,弃去上层清液,重复3次;收集沉淀烘干即得黄芪药渣.
1.2.2 醇不溶性残渣(A.I.R)粉末的制备 参照文献[8]方法,取黄芪药渣20 g,加入600 mL乙醇溶液(体积分数80%),于60℃水浴加热1 h,离心,弃去上层清液,重复多次,至上层清液无色;向沉淀中加入丙酮溶液(料液质量比1∶10),静置,弃去上层清液,沉淀烘干;在烘干后的粉末中加猪胰腺(料液质量比1∶100,50 mmol/L Tris⁃HCl,pH=7.0),置于摇床上孵育12 h(37℃,200 r/min),离心,弃去上层清液;用水清洗沉淀后,加入丙酮溶液(料液质量比1∶10),静置,弃去上层清液,沉淀烘干,得A.I.R粉末.
1.2.3 AXs的提取 参照文献[9]方法,称取A.I.R粉末20 g,加入400 mL 0.6%(质量分数)亚氯酸钠溶液,用冰醋酸调节pH=4.2~4.7,封口,置于75℃水浴中;每隔20 min手动摇动1次;1 h后继续加入0.6%亚氯酸钠溶液,并用冰醋酸调节pH=4.2~4.7;重复上述水解过程1 h.反应完成后,使体系温度迅速降低.离心、弃去上层清液后,用蒸馏水冲洗沉淀至中性,烘干得黄芪综纤维素.
[10]方法,称取黄芪综纤维素10 g,加入500 mL果胶酶[3 U/(mmol·L-1),乙酸钠50 mmol/L,pH=5.0],置于摇床上孵育24 h(24℃,200 r/min),离心,弃去上层清液,重复3次,用500 mL水清洗沉淀3次.向沉淀中加500 mL Na2CO3(50 mmol/L)和乙二醇二乙醚二胺四乙酸(5 mmol/L)的混合溶液,摇床孵育24 h(24℃,200 r/min),离心,弃去上层清液,重复3次,用500 mL水清洗沉淀3次.
向上述所得沉淀中加500 mL KOH(1 mol/L)和NaBH4(质量分数1%)的混合溶液,置于摇床上孵育12 h(24℃,200 r/min),离心,收集上层清液,重复3次,用500 mL水清洗沉淀3次,并将上层清液合并后于4℃保存,得黄芪半纤维素组分Ⅰ.
向上述半纤维素组分溶液中滴加冰醋酸至pH=5.0,透析,浓缩冻干,得半纤维素组分Ⅰ的多糖,标记为AX⁃Ⅰ.
1.3 Sevage法除蛋白及粗多糖分离纯化
利用Sevage法[11]对AX⁃Ⅰ进行脱蛋白处理.
[12]方法,将除蛋白后的AX⁃Ⅰ用DEAE⁃纤维素⁃52色谱柱和Sephacryl S⁃400 HR凝胶柱分离纯化,得纯化组分AX⁃Ⅰ⁃1,并用高效凝胶渗透色谱检验该多糖的纯度.
1.4 AX⁃Ⅰ⁃1的理化性质及结构分析
1.4.1 高效凝胶渗透色谱法测定相对分子量 取标准品Dextran 4320,Dextran 12600,Dextran 73800,Dextran 110000,Dextran 289000和Dextran 496000,配制成2 mg/mL的标准溶液用于高效凝胶色谱分析,每针进样20 μL,流动相为娃哈哈纯净水,流速为0.3 mL/min,柱温35℃,凝胶柱为TSKgel G4000PWxl(10)(7.8 mm×300 mm),示差折光检测器温度34℃,测定各标准品的保留时间.以保留时间(tR)为横坐标,分子量的对数lgMw为纵坐标,绘制标准曲线.
将多糖AX⁃Ⅰ⁃1配制成浓度为2 mg/mL的样品溶液.将测得的保留时间带入标准回归方程计算其分子量.
1.4.2 红外光谱分析 采用KBr压片法[13]测定样品的红外光谱(IR),扫描范围为4000~400 cm-1.
1.4.3 GC⁃MS法测定单糖组成 参照文献[14]方法,称取5 mg多糖AX⁃Ⅰ⁃1,加入1.5 mL三氟乙酸(2 mol/L),密封后于110℃油浴水解3 h,空气吹干后加入1 mL甲醇,重复3次,得到水解产物,溶于内标核糖醇溶液(0.1 mg/mL)中.加入NaHB4的DMSO溶液(质量分数2%),置于摇床上孵育90 min(40℃,200 r/min),用冰乙酸调pH至中性.加入500 μL醋酸酐和50 μL 1⁃甲基咪唑,反应10 min.加入5 mL蒸馏水终止反应,用二氯甲烷萃取2次,合并有机相,吹干后,复溶于二氯甲烷中,用0.22 μm有机滤膜过滤,取2 μL滤液注入GC⁃MS.
气相色谱条件:DB⁃5MS毛细管柱(30 m×0.5 cm×0.25 μm);载气为高纯氦气;流速1.0 mL/min;分流比10∶1;进样口温度220℃.程序升温:起始温度100℃,以5℃/min升至180℃,保持1 min;以1℃/min升至190℃,保持2 min;以30℃/min升至220℃,保持2 min;以1℃/min升至230℃,保持2 min;以20℃/min升至280℃,保持10 min.
质谱条件:Xcalibur 2.0.7工作站,电子轰击(EI)离子源,离子源温度220℃,传输线温度250℃,扫描模式Full Scan,扫描范围m/z 45~550.
1.4.4 甲基化分析 参照文献[15]方法,称取5 mg样品,加入3 mL无水二甲基亚砜,充入氩气,超声至样品溶解.称取NaH粉末50 mg,加入5 mL无水二甲基亚砜,充入氩气,磁力搅拌下于60℃反应至不再产生氢气为止.
将上述2种溶液合并,在氩气保护下超声1 h,冰浴冷却,避光加入1 mL CH3I,在氩气保护下,超声30 min,温度约为20℃,重复3次.加入5 mL Na2S2O3水溶液(4 mmol/L),再向反应液中加入5 mL CHCl3萃取,吸出下层,重复5次;合并下层溶液,加5 mL蒸馏水,萃取,弃去上层清液,重复5次.向氯仿相中加入无水Na2SO4,过滤,氮气吹干.用红外光谱检测3400 cm-1处无吸收峰,即为甲基化完全.加入2 mL三氟乙酸(2 mol/L),密封后于120℃水解90 min,空气吹干后加入甲醇,重复3次,得到水解产物.加入2 mL新配制的NaBD4水溶液(质量分数2%),置于摇床上孵育90 min(40℃,200 r/min),加冰乙酸调节pH至中性,吹干.加入500 μL醋酸酐和50 μL 1⁃甲基咪唑,反应10 min.加蒸馏水终止反应,用二氯甲烷萃取2次,合并有机相,吹干后,复溶于二氯甲烷中,用0.22 μm有机滤膜过滤,取2 μL注入GC⁃MS.
1.4.5 核磁共振分析 参照文献[16]方法,取干燥样品30 mg溶解在0.5 mL DMSO⁃d6中,于30℃测定1H NMR,13C NMR和2D NMR(HMBC和HMQC)谱.
1.5 黄芪药渣多糖AX⁃Ⅰ各组分的体外抗氧化作用
1.5.1 多糖AX⁃Ⅰ各组分对羟基自由基的清除作用 利用Fenton试剂[17]评价了黄芪药渣多糖AX⁃Ⅰ各组分对羟基自由基的清除作用.将多糖配成0.1,0.2,0.3,0.4和0.5 mg/mL的溶液.将1.0 mL硫酸亚铁溶液(9 mmol/L)、1.0 mL水杨酸⁃乙醇溶液(9 mmol/L)和1.0 mL多糖待测液混合均匀,加入1.0 mL过氧化氢溶液(8.8 mmol/L)启动反应,于37℃反应30 min,在510 nm波长下测定溶液的吸光度.阳性对照组为维生素C(Vc),空白对照组为蒸馏水.按下式计算清除率(Scavenging effect):

式中:A0为空白对照组的吸光度;Ai为多糖待测液组、阳性对照组的吸光度;Aj为1.0 mL水杨酸⁃乙醇溶液代替1.0 mL硫酸亚铁溶液作试剂空白的吸光度.
1.5.2 多糖AX⁃Ⅰ各组分对超氧阴离子的清除作用 采用邻苯三酚法测定了黄芪药渣多糖对O2-·自由基的清除作用[18].将多糖配成0.1,0.2,0.3,0.4和0.5 mg/mL的溶液.将5.0 mL Tris⁃HCl缓冲溶液(50 mmol/L,pH=8.2)和0.5 mL多糖溶液混合均匀,于37℃水浴预热10 min,加入1.0 mL邻苯三酚溶液(3.5 mmol/L),反应6 min后,迅速加入0.5 mL HCl(8 mmol/L)终止反应,于420 nm处测定吸光度.阳性对照组为Vc,空白对照组为蒸馏水.按下式计算清除率:
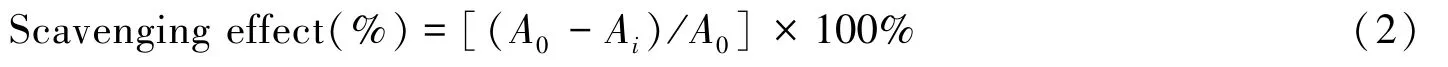
式中:A0为空白对照组吸光度;Ai为多糖待测液组、阳性对照组吸光度.
1.5.3 多糖AX⁃Ⅰ各组分对DPPH的清除作用 采用DPPH评价体系[19]研究了黄芪药渣多糖对DPPH自由基的清除作用.分别将各2.0 mL多糖溶液(0.1,0.2,0.3,0.4,0.5 mg/mL)和2.0 mL DPPH溶液(0.16 mmol/L)混合均匀,于25℃反应15 min,在517 nm处测定吸光度.阳性对照组为Vc,空白对照组为蒸馏水.按式(1)计算清除率,式中Aj为95%乙醇代替DPPH溶液作试剂空白的吸光度.
2 结果与讨论
2.1 黄芪药渣阿拉伯木聚糖的分离与纯化
根据硫酸⁃苯酚检测结果,从洗脱曲线(图1)可知,AX⁃Ⅰ经DEAE⁃纤维素52分离得到水洗脱组分AX⁃Ⅰ⁃1,NaCl(0.3 mol/L)洗脱组分AX⁃Ⅰ⁃2,NaOH(0.1 mol/L)洗脱组分AX⁃Ⅰ⁃3和NaOH(0.3 mol/L)洗脱组分AX⁃Ⅰ⁃4.

Fig.1 Anion⁃exchange chromatography of AX⁃Ⅰon DEAE⁃cellulose⁃52
从AX⁃Ⅰ中共分离得到4种组分,经真空冷冻干燥后均为白色粉末状的多糖.采用Sephacryl S⁃400 HR凝胶柱对多糖AX⁃Ⅰ⁃1进行纯化,经TSKgel G4000PWxl(10)凝胶渗透色谱柱测定,AX⁃Ⅰ⁃1的重均相对分子量为350000,且为单一对称峰,纯度较高.
2.2 AX⁃Ⅰ⁃1的红外光谱分析
在多糖AX⁃Ⅰ⁃1的红外光谱中,3407 cm-1处的强吸收峰为O—H的伸缩振动[20],2930和1426 cm-1处的吸收峰为C—H的伸缩振动和变角振动[21];1119和1045 cm-1处的吸收峰为吡喃糖环C—O—C的特征骨架振动[22];892 cm-1处的吸收峰为β型吡喃糖的特征吸收[23].由此可以初步判断,AX⁃Ⅰ⁃1的主链是β⁃吡喃型多糖.
2.3 AX⁃Ⅰ⁃1的单糖组成分析
采用GC⁃MS法分析单糖的组成,与6种单糖对照品的保留时间进行对比,发现AX⁃Ⅰ⁃1中含有鼠李糖(tR=24.73 min)、阿拉伯糖(tR=25.55 min)、木糖(tR=26.21 min)、甘露糖(tR=33.20 min)、葡萄糖(tR=33.37 min)和半乳糖(tR=33.76 min),其摩尔比为0.006∶14.113∶8.284∶0.116∶0.468∶1.
2.4 AX⁃Ⅰ⁃1的甲基化分析
通过对比AX⁃Ⅰ⁃1的GC⁃MS分析中碎片离子峰与美国佐治亚大学复合糖类研究中心(CCRC)数据库(https://www.ccrc.uga.edu/specdb/ms/pmaa/pframe.html)的数据(见表1)可知,AX⁃Ⅰ⁃1分支度较高,单糖残基以表1中的几种连接方式存在.
2.5 AX⁃Ⅰ⁃1的核磁共振分析
根据单糖组成分析、甲基化分析结果以及文献[24]报道的糖基的化学位移,对AX⁃Ⅰ⁃1在1D NMR(1H NMR,13C NMR)和2D NMR[HMBC(图2),HMQC(图3)]中的信号进行了归属.根据文献[24]报道的糖基化学位移,将糖基的端基质子信号进行归属并将其分别标记为A,B,C,D,E,F,G,H.在δ 1.24处的弱吸收峰是由α⁃L⁃Rhap中甲基产生的[25],但在13C NMR中δ 18.0~20.0处未发现甲基的吸收峰,这可能是由于α⁃L⁃Rhap的含量太小所致,由单糖组成分析结果也可发现Rha的含量很小,各糖基中不同的碳氢化学位移列于表2.

Table 1 Partially O⁃methylated alditol acetates of AX⁃Ⅰ⁃1

Fig.2 HMBC spectrum of AX⁃Ⅰ⁃1

Fig.3 HMQC spectrum of AX⁃Ⅰ⁃1

Table 2 Chemical shifts for the resonances of glycosyl residues of AX⁃Ⅰ⁃1 in1H and13C NMR spectra

Fig.4 Hypothetical structure of the repeat unit of AX⁃Ⅰ⁃1
根据甲基化分析结果和HMBC谱图中的相关峰对糖基的连接顺序进行了分析.在HMBC谱图中,相关峰δ 5.00/80.6,δ 5.00与F的1⁃H的δ 5.01最接近,δ 80.6与D的C2的δ 80.9最接近,因此F可能通过(1→2)苷键与D相连.在相关峰δ 4.90/83.8中,δ 4.90属于D的1⁃H,δ 83.8属于B的C3,因此D通过(1→3)苷键与B相连.在相关峰δ 4.86/84.5中,δ 4.86属于E的1⁃H,δ 84.5与B的C3最接近,因此E通过(1→3)苷键与B相连.在相关峰δ 4.83/84.3中,δ 4.83与G,H的1⁃H接近,δ 84.3与B的C3最接近,因此G和H通过(1→3)苷键与B相连.在相关峰δ 4.83/81.3中,δ 4.83与G和H的1⁃H接近,δ 81.3与F的C2及C的C4接近,因此G和H分别通过(1→2)苷键与F相连,通过(1→4)苷键与C相连.在相关峰δ 4.77/81.6中,δ 4.77属于C的1⁃H,δ 81.6与B的C4相近,因此C通过(1→4)苷键与B相连.在相关峰δ 4.90/83.0中,δ 4.90属于D的1⁃H,δ 83.0与B的C3最接近,因此D通过(1→3)苷键与B相连.在相关峰δ 4.26/73.5中,δ 4.26属于B的1⁃H,δ 73.5与C,F和H的C3接近,因此B可能与C,F和H通过(1→3)苷键相连.在相关峰δ 3.04/102.4中,δ 3.04属于C的2⁃H,δ 102.4与A,B,G和H的C1接近,因此A,B,G和H可能通过(1→2)苷键与C相连.综合单糖组成分析、甲基化分析和核磁共振分析的结果,推测AX⁃Ⅰ⁃1可能的结构如图4所示.其中,Ara和Xyl构成主链,主链构型为β型与红外图谱分析结果相符,其它糖组成支链.
2.6 黄芪药渣多糖AX⁃Ⅰ各组分体外抗氧化作用分析
2.6.1 多糖AX⁃Ⅰ各组分对羟基自由基的清除作用 羟基自由基是氧化性最强的活性氧自由基,通过考察黄芪药渣中多糖对羟自由基的清除能力,评价了其抗氧化性的强弱.图5示出了4个待测样品及Vc在不同浓度下对羟基自由基的清除作用,可见样品浓度为0.1~0.5 mg/mL时,各组分对羟基自由基的清除作用与其样品浓度不具有量效关系,清除能力不随浓度的变化而变化.各组分多糖对羟基自由基的清除能力较差,多糖AX⁃Ⅰ⁃2,AX⁃Ⅰ⁃3和AX⁃Ⅰ⁃4对羟基自由基的清除率均约为5%.

Fig.5 Hydroxyl radical scavenging assay

Fig.6 Superoxide radical scavenging assay
2.6.2 多糖AX⁃Ⅰ各组分对超氧阴离子的清除作用 邻苯三酚在碱性条件下可自氧化形成中间产物超氧阴离子自由基,此自由基能促进邻苯三酚自氧化,因此通过测定某物质对邻苯三酚自氧化的抑制作用,即可表征其对超氧阴离子自由基清除作用.由图6可知,当样品浓度为0.1~0.5 mg/mL时,多糖AX⁃Ⅰ⁃2,AX⁃Ⅰ⁃3和AX⁃Ⅰ⁃4对超氧阴离子的清除作用与浓度无正相关性.多糖AX⁃Ⅰ⁃4对超氧阴离子的清除率为40%.当浓度为0.1 mg/mL时,多糖AX⁃Ⅰ⁃2,AX⁃Ⅰ⁃3和AX⁃Ⅰ⁃4对超氧阴离子的清除率是阳性对照Vc的2倍,质量浓度为0.2 mg/mL时,多糖AX⁃Ⅰ⁃3和AX⁃Ⅰ⁃4对超氧阴离子的清除率略高于阳性对照Vc.
2.6.3 多糖AX⁃Ⅰ各组分对DPPH的清除作用DPPH自由基是一种稳定的有机自由基,广泛用于体外抗氧化实验,样品清除DPPH自由基的能力可反映其抗氧化活性的高低.由图7可知,当样品浓度为0.1~0.5 mg/mL时,多糖AX⁃Ⅰ⁃2,AX⁃Ⅰ⁃3和AX⁃Ⅰ⁃4对DPPH的清除率约为30%.多糖AX⁃Ⅰ⁃1对DPPH的清除作用较差,清除率约为5%.

Fig.7 DPPH radical scavenging assay
2.7 黄芪多糖和黄芪药渣多糖的差异性
黄芪多糖为细胞质可溶性多糖,目前已分离纯化出十几种不同结构的黄芪多糖[26].王莹等[27]纯化出的一种黄芪多糖的主要连接方式是α⁃D⁃(1→4)⁃葡聚糖,在每25个葡萄糖中有1个6⁃O位上的分支.方圣鼎等[28]从蒙古黄芪中得3种黄芪多糖Ⅰ,Ⅱ及Ⅲ;经鉴定黄芪多糖Ⅰ是杂多糖,由D⁃葡萄糖、D⁃半乳糖和L⁃阿拉伯糖组成;黄芪多糖Ⅱ和Ⅲ均为D⁃葡聚糖.陈启荣等[29]从膜荚黄芪水提液中得到一种α⁃(1→4)葡聚糖.由此可见,黄芪多糖多为葡聚糖,少数为杂多糖且所含单糖种类较少.黄芪药渣多糖为细胞壁结构性多糖,目前该类多糖的结构未见报道.本课题组[7]从黄芪药渣中用KOH(1和4 mol/L)提获得的粗多糖均为杂多糖,经进一步分离纯化得到8个组分;并发现黄芪药渣半纤维素多糖的主要成分是阿拉伯糖和木糖,还含有少量鼠李糖、甘露糖、葡萄糖和半乳糖,单糖组成与黄芪多糖差异较大.本研究中纯化出的黄芪药渣多糖AX⁃Ⅰ⁃1为杂多糖,含有6种单糖,比黄芪多糖中单糖种类多.AX⁃Ⅰ⁃1中单糖的连接位点较多,分支度高,空间结构比黄芪多糖复杂.
黄芪主要调节机体的免疫功能,临床上用于抗癌、防癌及消除慢性炎症等疾病,而多糖类是黄芪发挥免疫调节活性的主要物质.研究[30]表明,麦麸中半纤维素阿拉伯木聚糖具有较强的抗肿瘤活性,可同时提高机体特异性和非特异性免疫系统反应.通过增加延迟超敏反应、增长脾细胞寿命、增加自然杀伤细胞和巨噬细胞等的活力和数量,显著地抑制肿瘤生长,且经大鼠长期毒性实验显示,阿拉伯木聚糖无毒副作用,大鼠口服半数致死量(LD50)>36 g/kg.
3 结 论
从黄芪药渣中分离纯化出半纤维素组分AX⁃Ⅰ⁃1,AX⁃Ⅰ⁃2,AX⁃Ⅰ⁃3和AX⁃Ⅰ⁃4,研究了AX⁃Ⅰ⁃1的理化性质、化学结构及体外抗氧化活性.结果表明,AX⁃Ⅰ⁃1是一种杂多糖,含有鼠李糖、阿拉伯糖、木糖、甘露糖、葡萄糖和半乳糖(摩尔比为0.006∶14.113∶8.284∶0.116∶0.468∶1).该组分的主链是β⁃吡喃型多糖,高度分支化,含有的连接方式为L⁃Rhap⁃(1→,→3,4)⁃L⁃Arap⁃(1→,→2,3,4)⁃D⁃Xylp⁃(1→,→2)⁃D⁃Manp⁃(1→,D⁃Glcp⁃(1→,→2,3,6)⁃D⁃Glcp⁃(1→,→2,3)⁃D⁃Galp⁃(1→和→2)⁃D⁃Galp⁃(1→.4种半纤维组分对羟基自由基的清除率为5%.当浓度为0.1 mg/mL时,多糖AX⁃Ⅰ⁃2,AX⁃Ⅰ⁃3和AX⁃Ⅰ⁃4对超氧阴离子的清除率是阳性对照Vc的2倍;当浓度为0.2 mg/mL时,多糖AX⁃Ⅰ⁃3和AX⁃Ⅰ⁃4对超氧阴离子的清除率略高于阳性对照Vc.多糖AX⁃Ⅰ⁃2,AX⁃Ⅰ⁃3和AX⁃Ⅰ⁃4对DPPH的清除率为30%.本研究为黄芪药渣中半纤维素的开发利用奠定了基础.
[1] Gu G.G.,Sheng Nong’s Herbal Classi,People’s Medical Publishing House,Bejing,1995,11—15(顾观光.神农本草经,北京:人民卫生出版社,1995,11—15)
[2] Pan H.F.,Deng Q.D.,Zhao Z.M.,Lishizhen Medicine and Materia Medica Research,2011,22(8),2026—2027(潘华峰,邓乔丹,赵自明.时珍国医国药,2011,22(8),2026—2027)
[3] Jiang M.H.,Zhu L.,Jiang J.G.,Expert Opinion,2010,14(12),367—1402
[4] Zhou S.M.,Liu X.Z.,Guo Y.,Carbohydr.Polym.,2010,81,784—789
[5] Zhou Y.,Li S.,Qian Q.,Liu X.,Plant J.,2009,57,446—462
[6] Cao L.,Liu X.Z.,Qian T.X.,Sun G.B.,Guo Y.,Chang F.J.,Zhou S.M.,Sun X.B.,Inter.J.Biol.Macromol.,2011,48,160—164
[7] Hao X.,Li K.,Wang G.Z.,Journal of Shanxi Medical University,2016,47(4),338—343(郝霞,李科,王桂臻.山西医科大学学报,2016,47(4),338—343)
[8] Zhou Y.H.,Li S.B.,Qian Q.,Plant J.,2009,57,446—462
[9] Sun J.X.,Sun X.F.,Sun R.C.,Carbohydrate Polymers,2004,56,195—199
[10] Cheng X.Q.,Li K.,Chen X.M.,Journal of Beijing Forestry University,2012,34(5),44—49
[11] An X.J.,Feng L.,Song H.P.,Journal of Biology,2012,29(3),39—41(安晓娟,冯琳,宋红平.生物学杂志,2012,29(3),39—41)
[12] Liu Y.,Yin L.,Gong G.P.,Peng Y.F.,Huang L.J.,Wang Z.F.,Chem.J.Chinese Universities,2016,37(2),261—268(刘洋,殷璐,龚桂萍,彭艺芳,黄琳娟,王仲孚.高等学校化学学报,2016,37(2),261—268)
[13] Mouna S.,Hager L.,Mohamed D.,Santiageo G.,Chem.Res.Chinese Universities,2015,31(1),16—20
[14] Gao F.R.,Li K.,Hao X.,Chinese Traditional and Herbal Drugs,2015,46(14),2134—2142(高凡茸,李科,郝霞.中草药,2015,46(14),2134—2142)
[15] Ciucan I.,Kerek F.,Carbohydr.Res.,1984,131(2),209—217
[16] Tang X.B.,Li Z.H.,Li Y.H.,Liu W.,Yu P.,Li L.X.,Guo Y.,Chem.Res.Chinese Universities,2015,31(1),71—77
[17] Ma J.B.,Shen Y.S.,Li S.,Food Chemistry,2008,27,380—382
[18] Gregory P.,Food Chemistry,2015,169,430—438
[19] Sheng J.W.,Sun Y.L.,Carbohydrate Polymers,2014,108,41—45
[20] Chaikumpollert O.,Methacanon P.,Suchiva K.,Carbohydrate Polymers,2004,57(2),191—196
[21] Roy A.K.,Sen S.K.,Bag S.C.,Journal of Applied Polymer Science,1991,42(11),2943—2950
[22] Sun R.C.,Sun X.F.,Carbohydrate Polymers,2002,49(4),415—423
[23] Sun R.C.,Lawther J.M.,Banks W.B.,Carbohydrate Polymers,1996,29(4),325—331
[24] Pu X.Y.,Ma X.L.,Liu L.,Carbohydrate Polymers,2016,137,154—164
[25] Leone S.,Molinaro A.,Carbohydrate Research,2007,342,1514—1518
[26] Ji Y.H.,Zhang M.S.,Xu L.,Journal of Changchun University of Traditional Chinese Medicine,1999,15(3),62—63(纪耀华,张明淑,徐力.长春中医学院学报,1999,15(3),62—63)
[27] Wang Y.,Zhao Y.M.,Zhang Q.F.,Chinese Traditional and Herbal Drugs,2001,32(11),962—964(王莹,赵毅民,张起凤.中草药,2001,32(11),962—964)
[28] Fang S.D.,Xu X.Y.,Ye C.Q.,Chinese J.Org.Chem.,1982,2,26—31(方圣鼎,徐小异,叶纯渠.有机化学,1982,2,26—31)
[29] Chen Q.R.,Lin Z.H.,Ding L.X.,Chinese Journal of Modern Applied Pharmacy,1985,2(6),11—13(陈启荣,林振华,丁禄霞.中国现代应用药学,1985,2(6),1113—1115)
[30] Ghoneum M.,Gollapudi S.,Cancer Lett.,2013,10(201),41—49
(Ed.:P,H,D,K)
†Supported by the National Natural Science Foundation of China(No.31300278),the Science and Technology Research Program Fund for Shanxi Province,China(No.20150313004⁃5)and the Ministry of Education PhD New Teacher Fund,China(No.20131401120006).
Extraction,Separation,Structural Analysis and Antioxidant Activity in vitro of Arabinoxylans(AX⁃Ⅰ⁃1)from the Residue of Astragalus Root†
LIU Lei1,2,3,LI Ke2,3∗,HAO Xia1,2,3,WANG Guizhen1,2,3,QIN Xuemei2,3,DU Guanhua4,ZHANG Xiang5
(1.College of Chemistry and Chemical Engineering,2.Modern Research Center for Traditional Chinese Medicine,Shanxi University,Taiyuan 030006,China;3.The Efficacy of Chinese Medicine Material and New Drug Key Lab of Shanxi Province,Taiyuan 030006,China;4.Institute of Materia Medica,Chinese Academy of Medical Sciences&Peking Union Medical College,Beijing 100050,China;5.Medical College,University of Louisville,Louisville 40201,USA)
Four fractions(AX⁃Ⅰ⁃1,AX⁃Ⅰ⁃2,AX⁃Ⅰ⁃3 and AX⁃Ⅰ⁃4)from the residue of Astragalus root were extracted by sequential chemical extraction,and purified by DEAE⁃Cellulose 52 anion⁃exchange and Sepharcryl S⁃400 high resolution gel⁃permeation chromatography.A combination of chemical and instrumental analysis was performed to investigate the structural characterization and antioxidant activity of AX⁃Ⅰ⁃1.The re⁃sults demonstrated that AX⁃Ⅰ⁃1 was mainly composed of rhamnose,arabinose,xylose,mannose,glucose and galactose,and the molar ratio was 0.006∶14.113∶8.284∶0.116∶0.468∶1.Backbone of AX⁃Ⅰ⁃1 composed of arabinose and xylose by β⁃(1→2),β⁃(1→3)and β⁃(1→4)glycosidic bond.The branches were composed of(1→4)⁃linked⁃βArap,(1→3)⁃linked⁃βGalp,(1→2)⁃linked⁃βManp,and the terminal residues were αRhap,βGclp,βGalp.The four fractions had a certain scavenging effect on hydroxyl radical,superoxide anion radical and DPPH radical.When the concentration is 0.1 mg/mL,scavenging rate of AX⁃Ⅰ⁃2,AX⁃Ⅰ⁃3 and AX⁃Ⅰ⁃4 on superoxide anion is about 2 times higher than the positive control vitamin C.
Residue of astragalus root;Arabinoxylan;Purification;Structural analysis;Antioxidant activity
O629.12
A
10.7503/cjcu20160500
2016⁃07⁃13.网络出版日期:2016⁃11⁃11.
国家自然科学基金(批准号:31300278)、山西省科技攻关计划(批准号:20150313004⁃5)和教育部博士点新教师类基金(20131401120006)资助.
联系人简介:李 科,男,博士,副教授,主要从事糖化学与糖生物学方面的研究.E⁃mail:like@sxu.edu.cn
