环糊精包结谷胱甘肽的机理
2016-11-02邵学广蔡文生
沈 文, 邵学广,2, 蔡文生
(1. 南开大学化学学院分析科学研究中心, 天津市生物传感与分子识别重点实验室,天津化学化工协同创新中心, 天津 300071;2. 南开大学药物化学生物学国家重点实验室, 天津 300071)
环糊精包结谷胱甘肽的机理
沈文1, 邵学广1,2, 蔡文生1
(1. 南开大学化学学院分析科学研究中心, 天津市生物传感与分子识别重点实验室,天津化学化工协同创新中心, 天津 300071;2. 南开大学药物化学生物学国家重点实验室, 天津 300071)
使用分子动力学模拟结合自由能计算的方法在原子水平上研究了谷胱甘肽与α-,β-和γ-环糊精的包结模式, 计算了谷胱甘肽与3种环糊精之间6种可能包结过程的自由能变化. 结果表明, 谷胱甘肽的谷氨酸残基从α-环糊精大口端进入空腔最终形成的包结复合物最稳定; 在该复合物中, 谷氨酸残基的亚甲基链部分被完全包结在疏水空腔中, 其氨基与羧基位于与α-环糊精的小口端, 并与环糊精的伯羟基形成了氢键, 同时半胱氨酸中的巯基位于环糊精的大口端, 得到了有效的保护. 因此, 疏水相互作用和氢键相互作用构成了包结的主要驱动力.β-环糊精的优势包结模式与α-环糊精类似, 但形成复合物的稳定性次之, 而γ-环糊精由于空腔较大, 优势的包结模式是谷氨酸残基从γ-环糊精小口端进入空腔, 但所形成的复合物结构的稳定性最弱.
谷胱甘肽; 环糊精; 分子动力学模拟; 自由能计算
谷胱甘肽是一种由谷氨酸、 半胱氨酸与甘氨酸组成的天然活性肽[1,2]. 谷胱甘肽通常含有还原型谷胱甘肽(GSH)和氧化型谷胱甘肽(GSSG)两种存在形式. 通常, 只有GSH才具有生物活性, 因为GSH中的半胱氨酸侧链基团上的巯基可以保护重要的酶蛋白不被氧化, 从而保证酶的生理活性, 如图1(A)所示. 另一方面, 巯基与体内的自由基结合,自由基将还原为容易代谢的酸类物质, 加速自由基的排泄, 从而减轻自由基对重要脏器的损害[1]. 因此GSH作为抗氧化剂, 在人体内不仅可以保护重要蛋白质不被氧化, 还可以清除人体新陈代谢过程中产生的自由基, 所以GSH被制备成口服液以作为保健品. 但是GSH的巯基在水溶液和空气中极易被氧化, 成为GSSH, 从而失去生理活性, 人们希望利用合适的包结剂对GSH进行包结以保证巯基不被氧化, 从而提高谷胱甘肽的生物利用度. Alonso等[3]实验证实α-环糊精(CD)与GSH能够通过包结形成稳定的复合物, 该复合物在25~37 ℃下稳定存在, 然而实验上并未给出环糊精与谷胱甘肽的包结机理以及包结复合物的结构细节. 另外, 更大尺寸的β-和γ-CD对于GSH是否具有更高的结合能力, 目前并没有系统的研究[4~7].

Fig.1 Initial structures of GSH(A) and α-CD(B)
本文采用分子动力学(MD)模拟结合自由能计算的方法[8,9]研究了α-CD与GSH所有可能的包结模式以及形成稳定复合物的过程, 从原子水平上对包结结构和包结机理进行了探索, 并进一步研究了β-和γ-CD对GSH的包结过程, 比较不同尺寸的CD对GSH的包结能力, 进一步揭示包结驱动力和包结机理, 为设计基于环糊精的谷胱甘肽的包结剂提供了理论指导.
1 理论和方法
1.1模型建立
GSH的初始构象取自蛋白质晶体结构数据库(PDB code: 4ZBA),α-,β-及γ-CD的初始构象来自于其三维晶体结构[10~12](图1). 由于GSH的三叉结构, 3个氨基酸均有可能被CD的空腔包结, 即GSH含有3个可能的包结位点, 分别为甘氨酸残基(Gly), 谷氨酸残基(Glu)和半胱氨酸残基(Cys). 而每一种包结又可能有2种取向, 即从CD的大口端或小口端进入其空腔, 如图2所示. 本文对α,β,γ3种CD共18种可能的包结模式进行了模拟计算, 如表1所示. 首先建立每一种包结模式的初始模型, 进行能量最小化后分别置于带有周期性边界条件的水立方盒子中, 水盒子边缘与复合物任意一个原子的距离至少为1.5 nm. 在进行自由能计算时, 旋转环糊精使其空腔的主轴与Z轴平行.

Fig.2 Initial spacial arrangements of molecular systems with two orientations of GSH towards the CDs(A) Orien(Ⅰ); (B) Orien(Ⅱ). ξ: Projection onto the Z axis of the distance between the center of mass of Glu and that of CD.

SystemMethodNumberofatomsTime/nsGSH:α-CD[Gly(Ⅰ)]aMD724310GSH:α-CD[Gly(Ⅱ)]bMD714710GSH:α-CD[Glu(Ⅰ)]cMD+ABF724310+100GSH:α-CD[Glu(Ⅱ)]dMD+ABF757910+100GSH:α-CD[Cys(Ⅰ)]eMD709910GSH:α-CD[Cys(Ⅱ)]fMD722810GSH:β-CD[Gly(Ⅰ)]aMD754610GSH:β-CD[Gly(Ⅱ)]bMD775310GSH:β-CD[Glu(Ⅰ)]cMD+ABF727010+100GSH:β-CD[Glu(Ⅱ)]dMD+ABF777710+100GSH:β-CD[Cys(Ⅰ)]eMD716810GSH:β-CD[Cys(Ⅱ)]fMD775310GSH:γ-CD[Gly(Ⅰ)]aMD745010GSH:γ-CD[Gly(Ⅱ)]bMD800810GSH:γ-CD[Glu(Ⅰ)]cMD+ABF745010+100GSH:γ-CD[Glu(Ⅱ)]dMD+ABF800810+100GSH:γ-CD[Cys(Ⅰ)]eMD778310GSH:γ-CD[Cys(Ⅱ)]fMD766610
a. Inclusion of the Gly residue of GSH into CDs with Orien(Ⅰ);b. inclusion of the Gly residue of GSH into CDs with Orien(Ⅱ);c. inclusion of the Glu residue of GSH into CDs with Orien(Ⅰ);d. inclusion of the Glu residue of GSH into CDs with Orien(Ⅱ);e. inclusion of the Cys residue of GSH into CDs with Orien(Ⅰ);f. inclusion of the Cys residue of GSH into CDs with Orien(Ⅱ).
1.2模拟参数
采用NAMD 2.11软件进行MD模拟[13], 分别采用CHARMM36力场[14,15]和TIP3P模型[16]来描述复合物及水分子. 采用SHAKE/RATTLE算法[17,18]将非水分子中含有氢原子的共价键的长度限制在其平衡值, 采用SETTLE算法[19]保持水分子的刚性. 对运动方程积分的时间步长为2 fs, 利用Langevin动力学和Langevin活塞方法[20]将温度和压力分别控制在300 K和1.01325×105Pa. 范德华截断半径为1.4 nm, 长程静电相互作用采用粒子网格埃瓦尔德(PME)方法计算[21]. 对于每个体系, 先约束溶质, 对溶剂进行10000步优化, 然后去除所有约束, 将经过5000步的能量最小化和10 ns的MD模拟之后的结构作为自由能计算的初始结构. 采用VMD软件[21]进行轨迹分析和可视化.
1.3自由能计算
MD模拟的结果表明, 18种可能的包结模式中只有GSH的Glu进入CD空腔的包结模式是可能稳定存在的. 因此只考察Glu分别从CD大口端和小口端进入CD空腔的过程, 并计算相应包结过程的自由能变化. 模拟反应坐标ξ定义为Glu的质心(由Cα和侧链上3个碳原子的质心定义)与CD质心(由其6个糖苷键上的氧原子的质心定义)之间的距离在Z轴方向上的投影, 反应坐标初始值为-0.8 nm, 采用自适应偏置力计算方法(ABF)计算[22~28]自由能沿模拟反应坐标的变化(Free-energy profile), 也称为平均力势(PMF)曲线. 为了保持CD空腔的中心轴在模拟过程中与Z轴方向一致, 在CD糖苷氧上施加了418 kJ·mol-1·nm-2的谐振约束. 为了提高采样的均匀性与平均力的连续性, 路径被分成了长度为0.1 nm的连续窗口, 反应路径变化的步长为0.01 nm. 每个窗口至少进行15 ns的MD模拟.
2 结果与讨论
2.1包结模式的探索

Fig.3 Distance between the center of residues of GSH and CD as a function of simulation time(A) GSH:α-CD; (B) GSH: β-CD; (C) GSH: γ-CD. The Gly residue of GSH and CD with Orien(Ⅰ); the Gly residue of GSH and CD with Orien(Ⅱ); the Glu residue of GSH and CD with Orien(Ⅰ); the Glu residue of GSH and CD with Orien(Ⅱ); the Cys residue of GSH and CD with Orien(Ⅰ); the Cys residue of GSH and CD with Orien(Ⅱ).
对谷胱甘肽与3种环糊精的18种包结模式分别进行10 ns的MD模拟. 为了考察每种包结模式是否可能存在, 计算了GSH的包结残基与CD质心间距离随模拟时间的变化情况, 每种包结模式初始时, 质心间距离为0.2~0.5 nm, 结果如图3所示. 可见, 在GSH:α-CD体系中, 谷胱甘肽的Glu与α-CD间的距离无论以哪种取向, 在模拟时间内均始终保持稳定, 大约在0.25 nm左右, 表明Glu可以被α-CD稳定包结. 而Gly和Cys与α-CD质心间距离随着模拟进行逐渐增大且波动剧烈, 表明GSH已处于游离状态, Gly与Cys不能被α-CD包结.
在GSH:β-CD体系中, 3种残基与β-CD质心间距离随模拟时间的演化与GSH:α-CD体系相似, 表明Glu可以被β-CD包结. 值得一提的是, Gly和β-CD以Gly(Ⅱ)取向包结的模拟轨迹显示, 3 ns后Gly从β-CD的小口端穿越空腔到达大口端, 同时Glu从小口端进入并停留在CD空腔中, 最终形成稳定的包结结构. 综合6个模拟结果, 只有Glu可以被β-CD稳定包结.
GSH:γ-CD(Cys)的模拟结果与其它2种CD相应的模拟结果类似, 2 ns后Cys迅速脱离空腔, 不能形成稳定的复合物. GSH:γ-CD(Gly)的模拟结果显示, 由于γ-CD空腔足够大, GSH在CD腔内也很容易调整姿态, 最终形成的包结物结构与GSH:γ-CD(Glu)模拟得到的结构相似, 即Glu被完全包结, 而Gly位于CD的口端, 被部分包结.
综上, 对18种GSH:CD体系的MD模拟结果表明, 由于Cys残基最短, 不能深入CD的空腔, 因此, GSH:CD(Cys)包结模式是不存在的; 而Glu残基无论以何种取向均可进入3种CD的空腔并形成稳定的复合物结构; CD对Gly的包结略有不同, 在α-CD中, Gly迅速解离, 在β-CD中, 观察到其中一种取向的包结最终转变成了对Glu的包结, 而在γ-CD中, 2种取向的初始包结均转变成了对Glu的包结. 可见, 与Gly相比, CD与Glu残基的包结在能量上更具优势. 因此, 本文只对GSH:CD[Glu(Ⅰ, Ⅱ)]的包结过程进行自由能计算.
2.2自由能曲线
图4描述了谷氨酸残基以2种取向[Glu(Ⅰ)与Glu(Ⅱ)], 分别从距离环糊精质心外0.8 nm处进入环糊精空腔的自由能变化. 在GSH:α-CD体系中, Glu从α-CD大口端进入, 穿越空腔的自由能曲线, Glu(Ⅰ)取向有2个能量极小值点, 从小口端进入空腔, 即Glu(Ⅱ)取向, 也有2个能量极小值点, 2种取向下的第2个能量最低点的结构如图5(A)所示, 从自由能曲线可见, GSH从大口端进入空腔, 形成的复合物更加稳定. 但是在2种取向下, 形成最稳定的复合物结构前, 均经历了一个能垒最高点, 这是由于α-CD空腔较小导致的, 而Glu通过α-CD小口端时, 会导致一个较高的能垒, 这也解释了Glu(Ⅱ)比Glu(Ⅰ)更早到达能垒最高点的现象.

Fig.4 Free-energy profiles for the inclusion of the Glu of GSH into CDs in two orientations (A) GSH:α-CD; (B) GSH:β-CD; (C) GSH:γ-CD. a. Orien(Ⅰ); b. Orien(Ⅱ).
由图4(B)可见, 描述β-CD包结GSH的2种取向的自由能变化曲线上均仅含有一个能量最低点. 对比2种取向下的自由能曲线, 与GSH:α-CD类似, GSH从β-CD的大口端进入空腔[Glu(Ⅰ)]形成的复合物更稳定. 由于β-环糊精空腔较大, GSH的Glu通过β-CD小口端时无空间位阻, 故无能垒. 在GSH:γ-CD体系中, 与β-CD类似, 因为空腔的增大而未产生能垒, 通过观察图4的自由能曲线, GSH从γ-CD的小口端进入空腔[Glu(Ⅱ)]形成的复合物最稳定. 可见, 谷胱甘肽与3种环糊精分别以2种取向形成的6种复合物中, GSH从α-CD大口端进入[Glu(Ⅰ)]形成的复合物结构最稳定.
2.3包结复合物的结构和氢键分析
为了研究谷胱甘肽与环糊精复合物在全局能量最小点的稳定结构, 提取出全局能量最低点的代表结构, 如图5所示. 对应于图4中每个自由能变化曲线能量最低点处的代表性结构, 在图5(A1和A2)中, Glu从α-CD大口端进入[Glu(Ⅰ)], 头部穿越小口端并停留在小口端附近, 疏水链被完全包结在环糊精的空腔中, Cys的巯基位于大口端附近, 形成了可以保护巯基的复合物. 由图5可见, [Glu(Ⅱ)]所有复合物的巯基都裸露在空腔外, 可见, 谷胱甘肽的Glu从α-CD大口端进入空腔所形成的复合物不仅结构最稳定, 还起到了有效保护GSH巯基的作用.

Fig.5 Structures of the inclusion complexes of GSH with CD near the global minima of the PMFs in Fig.4(A1) GSH with α-CD, Orien(Ⅰ): ξ=-0.02 nm, d=0.05 nm, θ=14°; (A2) Orien(Ⅱ): ξ=-0.04 nm, d=0.22 nm, θ=12°; (B1) GSH with β-CD, Orien(Ⅰ): ξ=-0.30 nm, d=0.31 nm, θ=28°; (B2) Orien(Ⅱ): ξ=-0.18 nm, d=0.28 nm, θ=22°; (C1) GSH with γ-CD, Orien(Ⅰ): ξ=-0.10 nm, d=0.49 nm, θ=43°; (C2) Orien(Ⅱ): ξ=-0.05 nm, d=0.44 nm, θ=37°. ξ: Reaction coordinate in Fig.4; d: distance between the center of mass of Glu and the principal axis of CD; θ: angle between the side chain of Glu and the principal axis of CD.
为了研究Glu从α-CD大口端进入[Glu(Ⅰ)]形成复合物较为稳定的原因, 探讨了GSH与α-CD之间氢键的变化关系, 如图6所示. 图6(A)为GSH与α-CD沿反应路径的氢键数量变化趋势图. 对比GSH与α-CD自由能变化曲线可见, 在自由能最低点时, 氢键形成数目最多. 同时, 图6(B)为自由能最低点时GSH与α-CD之间形成的氢键示意图. 可见, 此包结模式最多可以形成3个氢键, 分别是α-CD小口端的羟基与Glu上的氨基和羧基形成2个氢键,α-CD大口端的羟基与Cys上的羧基形成第3个氢键. 氢键强有力的形成保证了包结模式的稳定性. 而第3个氢键的形成保证了Cys上的巯基可以被保护在α-CD空腔内.
对比图6(A)氢键数量变化趋势图与图4(A)GSH:α-CD体系的自由能变化曲线发现, 虽然在ξ为-0.2 nm处形成的氢键也较多, 但是此时的自由能曲线位置处于能量最高点, 针对此现象, 继续进行了环糊精变形程度的分析.
2.4环糊精形变程度分析
图7(A)为在GSH在穿梭过程中,α-CD小口端上的6个C原子所组成的六边形的面积随反应路径的变化趋势图; 图7(B)为在GSH穿梭过程中,α-CD中心处的6个糖苷键氧原子所形成的六边形的面积随反应路径的变化趋势图. 由图7(B)可知, 在ξ为-0.2 nm时,α-CD中心处的形变达到极大值, 主要是因为GSH上Glu的氨基与羧基位于环糊精中心处, 具有较大的空间位阻. 此时, 虽然环糊精与谷胱甘肽之间形成较多氢键, 但依然有较高的自由能能垒. 在ξ为0~0.3 nm之间时, 环糊精的小口端形变程度减小[见图7(A)], 但是由于甘氨酸残基进入环糊精空腔, 造成了新的空间位阻[见图7(B)], 因此导致自由能再次升高. 结合以上分析可见, 在ξ为-0.2 nm处虽然可以形成较多的氢键, 但是环糊精中心处形变程度较大, 所以形成了一个较高的能垒.

Fig.7 Fluctuation of the area of the central plane formed by the six primary side carbon atoms(A) and the six glycosidic oxygen atoms(B) of the α-CD in the course of the threading process
2.5GSH与α-CD之间的疏水相互作用

Fig.8 Variation of the solvent-accessible surface area(SASA) for the hydrophobic chain of the Glu as a function of the model reaction coordinate
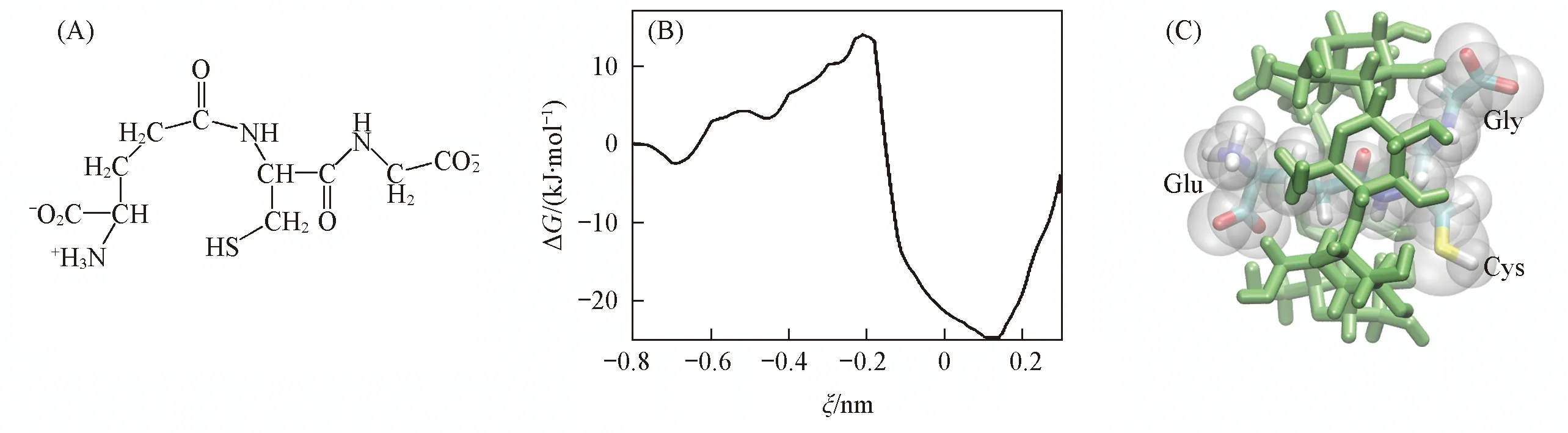
Fig.9 Structure of GSH-(A), free-energy profile for the inclusion of the Glu of GSH- into α-CD in Orien(I)(B) and structure of the inclusion complex of α-CD with GSH- near the global minima of the PMF(C)
为了研究GSH与α-CD之间的疏水相互作用, 探讨了Glu从α-CD大口端穿越空腔过程中, 谷胱甘肽Glu中亚甲基链的溶剂可及表面随反应坐标的变化趋势. 如图8所示, 即ξ在-0.1~0 nm时, 亚甲基链的溶剂可及表面最低, 此时恰好是自由能的最低点(图4). 此时, 亚甲基链被完全包结在α-CD的疏水空腔中, 亚甲基链与α-CD的疏水空腔之间有利的疏水相互作用, 不仅增强了包结复合物结构的稳定性, 也是促进GSH与α-CD包结的驱动力之一.
2.6GSH电离状态对包结结构的影响
GSH共有16种不同的电离状态, 而在生理环境下主要以GSH-的形式存在[29], 如图9(A)所示. 根据以上对α-CD与GSH各种可能包结模式的研究所得最优取向的包结过程进行自由能计算, 即计算Glu由环糊精大口端进入空腔[Orien(Ⅰ)]的自由能变化, 结果如图9(B)所示.
可见, 该曲线与GSH从α-CD大口端进入的自由能变化曲线的趋势基本一致, 但只有一个明显的稳定态, 对应的最稳定结构如图9(C)所示, 与图5(A)[Orien(Ⅰ)]的结构相似. 可见, GSH的这种电离状态对与α-CD形成的包结结构的影响不大.
3 结 论
利用分子动力学模拟结合自由能计算研究了谷胱甘肽与3种天然环糊精的包结行为, 得到了最稳定的包结结构, 揭示了相应的包结机理. 结果表明, GSH的Glu残基可以被环糊精空腔包结形成稳定的复合物, 但由于β-CD与γ-CD的空腔相对较大, Glu的体积较小, GSH与β-CD或γ-CD形成的复合物没有与α-CD形成的复合物稳定. 进一步分析了Glu与α-CD 2种不同取向包结过程的自由能变化以及包结过程中疏水和氢键相互作用, 结果表明, 当Glu从α-CD大口端进入空腔, 在疏水相互作用和静电相互作用的驱动下, 其疏水链被完全包结在环糊精空腔中, 同时GSH和α-CD之间形成多个氢键, 最终形成的复合物最稳定. 结构分析结果显示, 在该复合物中GSH上的巯基被α-CD的大口端包结, 从而得到有效的保护, 达到了防止其被氧化的目的. 进一步对GSH-与α-CD的包结进行了计算, 结果表明, 所形成的包结结构未受到GSH电离状态的明显影响, 说明α-环糊精是一个很好的谷胱甘肽包结剂. 本文的研究结果为设计新的谷胱甘肽包结剂提供了理论依据.
[ 1 ]Zetterstrom R., Eijkman C., Hopkins F. G.,ActaPaediatr., 2006, 95(11), 1331—1333
[ 2 ]Dixon D. P., Skipsey M., Grundy N. M.,PlantPhysiol., 2005, 138, 2233—2244
[ 3 ]Garcia-Fuentes M., Trapani A., Alonso M. J.,Eur.J.Pharm.Biopharm., 2006, 64(2), 146—153
[ 4 ]Song L. X., Guo Z. J.,ChineseJ.Inorg.Chem., 2001, 17(4), 457—470(宋乐新, 郭子建. 无机化学学报, 2001, 17(4), 457—470)
[ 5 ]You C.C., Liu Y.,Chem.J.ChineseUniversities, 2000, 21(2), 249—251(尤长城, 刘育. 高等学校化学学报, 2000, 21(2), 249—251)
[ 6 ]You C.C., Zhao Y. L., Liu Y.,Chem.J.ChineseUniversities, 2001, 22(2), 218—222(尤长城, 赵彦利, 刘育. 高等学校化学学报, 2001, 22(2), 218—222)
[ 7 ]Song L. X.,Wang H. M., Yang Y.,ActaChim.Sinica, 2007, 65(16), 1593—1599(宋乐新, 王海名, 杨燕. 化学学报, 2007, 65(16), 1593—1599)
[ 8 ]He J., Feng X. Z., Shao X. G., Cai W. S.,Chem.J.ChineseUniversities, 2015, 36(1), 110—115(何佳, 冯喜增, 邵学广, 蔡文生. 高等学校化学学报, 2015, 36(1), 110—115)
[ 9 ]Cai W. S., Sun T. T., Shao X. G., Chipot C.,J.Phys.Chem.B, 2009, 113(22), 7836—7843
[10]Harata K.,Bull.Chem.Soc.Jpn., 1975, 48(9), 2409—2413
[11]Lindner K., Saenger W.,CarbohydrRes., 1982, 99(2), 103—115
[12]Harata K.,Bull.Chem.Soc.Jpn., 1987, 60(8), 2763—2767
[13]Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kale L., Schulten K.,J.Comput.Chem., 2005, 26(16), 1781—1802
[14]MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiorkiewicz Kuczera J., Yin D., Karplus M.,J.Phys.Chem.B, 1998, 102(18), 3586—3616
[15]MacKerell A. D., Feig M., Brooks C. L.,J.Comput.Chem., 2004, 25(11), 1400—1415
[16]Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L.,J.Chem.Phys., 1983, 79(2), 926—935
[17]Ryckaert J. P., Ciccotti G., Berendsen H. J. C.,J.Comput.Phys., 1977, 23(3), 327—341
[18]Andersen H. C.,J.Comput.Phys., 1983, 52(1), 24—34
[19]Miyamoto S., Kollman P. A.,J.Comput.Chem., 1992, 13(8), 952—962
[20]Feller S. E., Zhang Y. H., Pastor R. W., Brooks B. R.,J.Chem.Phys., 1995, 103(11), 4613—4621
[21]Humphrey W., Dalke A., Schulten K.,J.Mol.Graphics, 1996, 14(1), 33—38
[22]Wang S. S., Liu P., Cai W. S., Shao X. G.,Chem.J.ChineseUniversities, 2015, 36(11), 2211—2219(汪双双, 刘鹏, 蔡文生, 邵学广. 高等学校化学学报, 2015, 36(11), 2211—2219)
[23]Darden T., York D., Pedersen L.,J.Chem.Phys., 1993, 98(12), 10089—10092
[24]Darve E., Pohorille A.,J.Chem.Phys., 2001, 115(20), 9169—9183
[25]Hénin J., Chipot C.,J.Chem.Phys., 2004, 121(7), 2904—2914
[26]Rodríguez-Gómez D., Darve E., Pohorille A.,J.Chem.Phys., 2004, 120(8), 3563—3578
[27]Chipot C., Hénin J.,J.Chem.Phys., 2005, 123(24), 244906
[28]Hénin J., Fiorin G., Chipot C., Klein M. L.,J.Chem.TheoryComput., 2010, 6(1), 35—47
[29]Lampela O., Juffer A. H., Rauk A.,J.Phys.Chem.A, 2003, 107(43), 9208—9220
(Ed.: Y, Z, S)
† Supported by the National Natural Science Foundation of China(No.21373117).
Inclusion Mechanism of Cyclodextrins with Glutathione†
SHEN Wen1, SHAO Xueguang1,2, CAI Wensheng1*
(1.ResearchCenterforAnalyticalSciences,CollegeofChemistry,TianjinKeyLaboratoryofBiosensingandMolecularRecognition,CollaborativeInnovationCenterofChemicalScienceandEngineering(Tianjin),NankaiUniversity,Tianjin300071,China;2.StateKeyLaboratoryofMedicinalChemicalBiology,NankaiUniversity,Tianjin300071,China)
By using molecular dynamics simulations combined with free energy calculations, the inclusion modes ofα-,β-, andγ-cyclodextrins(CDs) with glutathione(GSH) in an aqueous environment were investigated at the atomic level. The free-energy changes for the six possible inclusion processes of the three types of CDs with GSH were calculated. The results show that the inclusion complex formed by GSH andα-CD with the orientation that the glutamic acid(Glu) residue entering from the secondary rim of the CD is the most energetically favored, wherein the methylene chain of the Glu is completely buried in the cavity ofα-CD, and three hydrogen bonds are formed betweenα-CD and GSH. It is worth noting that in this most stable complex structure, the sulfhydryl group of GSH is included at the secondary rim, thereby being protected byα-CD. Moreover, hydrophobic and hydrogen-bonding interactions constitute the main driving force responsible for the formation of the host-guest complex. The favorable inclusion mode of GSH withβ-CD is similar to that withα-CD, but the corresponding complex of the former is less stable than that of the latter. On the contrary, forγ-CD, the favorable orientation is that the Glu residue enters the cavity from the primary side of the CD. From the free-energy calculations reported herein, the relative binding affinity with GSH follows the ranking orderα-CD>β-CD>γ-CD.
Glutathione; Cyclodextrin; Molecular dynamics simulation; Free-energy calculation
10.7503/cjcu20160375
2016-05-25. 网络出版日期: 2016-09-23.
国家自然科学基金(批准号: 21373117)资助.
O641
A
联系人简介: 蔡文生, 女, 博士, 教授, 博士生导师, 主要从事分子模拟和化学信息学研究. E-mail: wscai@nankai.edu.cn
