左心室受累的致心律失常心肌病研究进展
2016-10-27范思洋综述樊晓寒审校
范思洋综述,樊晓寒审校
左心室受累的致心律失常心肌病研究进展
范思洋综述,樊晓寒审校
目前在临床上发现致心律失常右心室心肌病中有许多合并累及左心室的病例,还有部分单纯累及左心室的致心律失常心肌病。左心室受累的致心律失常性心肌病流行病学研究不多,临床特征主要表现为左心室起源的室性心律失常,早期可能只有心电学的改变,左心室结构无明显异常,磁共振检查可见左心室延迟增强等影像学异常,临床预后比单纯累及右心室更差,心原性猝死发生率高。左心室受累的致心律失常心肌病的致病突变基因同样以编码桥粒蛋白的基因突变为主,但其不同于右心室心肌病(常见突变基因PKP2),DSP基因是其最常见的突变基因。
综述;心肌疾病;心律失常,心性
近年来越来越多的研究发现,不仅只有单纯致心律失常右心室心肌病(ARVC),还有同时或单纯累及左心室的致心律失常心肌病[1]。2011年由美国心律协会(HRS)和欧洲心律学会(EHRA)正式提出致心律失常心肌病(Arrhythmogenic Cardiomyopathy)的定义[2]。目前有学者将致心律失常性心肌病分为3个亚型,右心室型、左右心室型和左心室型[1]。本文就左心室受累的致心律失常心肌病(左心室型,左右心室型)的流行病学、临床特征和预后、遗传特征和致病基因等几个方面进行综述。
1 左心室受累的致心律失常心肌病的流行病学
致心律失常心肌病在西方人群中的发病率为1/2 000~1/5 000,是导致年龄≤35岁人群发生心原性猝死的重要病因,占65岁以下人群不明原因猝死10%以上[3]。早期的西方人群研究就发现,临床诊断为ARVC的患者中经尸检或影像学诊断确定的累及左心室的比例高达76%~84%[1,4]。1997年有研究在42例经过尸检或心脏移植后病检明确诊断的ARVC患者发现,有76%(32例)患者发现左心室有脂肪浸润,其中只有17例有左心室组织学和形态学的改变[4]。Sen-Chowdhry等[1]收集了200例临床诊断符合早期工作组共识(TF)诊断ARVC标准或修改后家族遗传性ARVC诊断标准的患者,依据心脏磁共振检查结果,有84%(168例)的患者有左心室受累的影像学异常,包括92%(155例)有左心室延迟增强,43%(72例)有左心室脂肪浸润,还有40%有左心室扩张,18%的患者有左心室射血分数下降。近年有一项多中心研究[5]对50例发生心脏性猝死、心室有纤维脂肪浸润的心肌病患者进行尸检,发现只有12%单纯累及右心室,50%患者的心肌纤维脂肪病变累及双心室,还有38%单纯累及左心室。该研究还发现累及不同心室的致心律失常心肌病的比例在不同种族中有差异。50例患者中黑人占50%,白人44%,亚洲人只有4%(2例)。在白人和黑人中,有50%左右的患者是累及双心室的心肌病,30%左右患者单纯累及左心室,只有10%左右的患者单纯累及右心室。而入选的2例亚洲患者1例累及双心室,1例单纯累及左心室。该研究亚洲患者入选少,似乎亚洲患者均累及左心室,但亚洲患者累及左心室的致心律失常心肌病的患病率还有待大样本研究提供确切的数据。
2 左心室受累的致心律失常心肌病的临床特征和预后
2.1累及左心室的致心律失常心肌病的临床、心电和影像学表现
左心室受累的致心律失常心肌病包括单纯累及左心室的左心室型(LDAC)和同时累及左右心室的左右心室型致心律失常心肌病。LDAC被认为在家族性致心律失常心肌病的疾病表达模式中占三分之一。相比较经典的ARVC,LDAC早期就有左心室病变,且主要累及左心室[1]。在病理上以左心室被纤维脂肪组织所替代为特征,在心原性猝死患者的尸检中被发现,左心室纤维脂肪组织经常以环形发生在心肌外1/3和室间隔的右心室侧[6]。LDAC的临床表现多样,如胸痛,持续心悸、劳力性呼吸困难、晕厥等左心室功能损伤的相关症状,少部分患者无明显症状[1,7]。心律失常类型主要为右束支阻滞形的频发室性早搏或室性心动过速[8,9]。心电图特征表现[1,7,10]为:侧壁导联(V5、V6、伴/不伴V4,I和aVL)或下壁导联(Ⅱ,Ⅲ,aVF)可出现T波倒置和epsilon波,可出现左束支阻滞,电轴左偏。在临床中,如果排除阻塞性冠状动脉疾病和左心室收缩功能障碍,这些常常是在临床上被忽视的心电图异常改变。经典ARVC主要是起源于非右心室流出道的左束支阻滞形频发室性早搏或室性心动过速,心电图特征是右心室前壁导联(V1~V3)出现室内传导阻滞、宽QRS波、T波倒置、epsilon波和ST段抬高等。另外这两种类型的致心律失常心肌病的信号平均心电图都可显示晚电位,都会偶尔出现室上性心动过速、心房颤动或心房扑动等。
左右心室型致心律失常心肌病的特征是早期左心室和右心室病变就平行发展,表现为左右心室均有扩张或收缩功能障碍。病理上左、右心室均有纤维脂肪替代,右心室和左心室的容量比为1左右[1,5]。临床上有左、右心室同时损伤的表现,如同时有左束支阻滞形和右束支阻滞形的室性心律失常,心电图上既有左心室侧壁和下壁导联的异常又有右心室前壁导联异常。左右心室型致心律失常心肌病也会偶尔出现室上性心动过速、心房颤动或心房扑动等。
病理学检查发现心室肌间脂肪纤维组织浸润是诊断ARVC的金标准。黄洁等[11]回顾分析了49例心脏移植患者,其中9例经病理证实为左心室受累为主的ARVC患者,而这些患者按照1994年McKenna等[12]制定的标准,仅1例可临床诊断为ARVC。但由于病理学检查取样为有创操作,临床上患者很难接受导致较少应用,主要于心脏移植后或尸检中行病理学检查[5]。
影像学检测是致心律失常心肌病诊断的重要依据。LDAC的超声心动图可表现为左心室射血分数轻微下降或正常,左心室舒张末容积增加和左心室室壁运动异常[1,7]。磁共振检查主要表现为左心室局限性扩张,室壁运动障碍,左心室室壁瘤,左心室致密化不全,左心室心外膜下或中层心肌延迟增强等[1,7]。而ARVC主要表现为右心室局限性扩张,右心室室壁运动障碍,右心室室壁瘤,特别是好发于由右心室流入道、流出道和心尖部组成的发育不良三角,还有异常的小梁形成,以及右心室延迟增强等[13]。另外,超过50%的LDAC患者都有室间隔的延迟增强,而经典ARVC没有此表现[1]。左心室延迟增强是间质纤维化或替代纤维化的表现。LDAC的典型左心室延迟增强部位多在左心室下壁和侧壁,并影响心肌的心外膜层和中层,呈圆周状分布,这与LDAC病理解剖上纤维脂肪替代形成的环形带是一致的[4]。在没有室壁运动障碍和没有左心室球形扩张或收缩功能障碍的患者中检测到左心室延迟增强,有助于LDAC的早期诊断。黄静涵等[14]入选从2008-08至2010-05在阜外医院诊断为ARVC的患者64例,其中48例(75%)磁共振检查阳性,4例磁共振检查累及左心室,但心电图未发现侧壁或下壁导联异常等典型累及左心室特征。有研究发现,延迟增强范围与心律失常事件发生具有相关性[7],可预测预后的危险程度。当临床上出现上述心律失常类型并伴有心电图、信号平均心电图以及超声磁共振等影像学特征,而且排除阻塞性冠状动脉疾病和左心室收缩功能障碍,应高度怀疑为LDAC[1,7]。
2.2左心室受累的致心律失常心肌病的预后
左心室受累的致心律失常心肌病的预后资料目前较少,但左心室的结构和功能决定了累及左心室的心肌病较单纯累及右心室更加预后不良[15]。左心室功能障碍增加恶性心律失常事件和猝死危险。左心室结构和功能受损容易导致体循环灌注不足继而导致心力衰竭[16]。有些LDAC在心律失常事件发生时,左心室结构和功能无异常,仅仅有心悸或晕厥的症状,在临床上容易被忽略,可因恶性心律失常事件而直接发生心原性猝死。目前预后方面无大规模临床试验数据,尚待进一步研究证实。
3 左心室受累的致心律失常心肌病的致病相关基因
目前已证实5个编码细胞桥粒蛋白的基因突变可导致传统定义的ARVC的表型出现,主要是PKP2,DSP,DSG2,DSC2,JUP,CTNNA3,TMEM43[17]基因。后期研究发现6个非桥粒基因突变也可导致ARVC的表型,包括RYR2, TGFB3,DES,LMNA,TTN,PLN[17]基因。累及左心室的致心律失常心肌病的致病基因国内外相关研究报道较少。目前基因突变筛查并不能作为临床诊断和分型的依据,只能作为临床诊断的补充提示。
表1列出近年来报道的通过家系研究发现的累及左心室致心律失常心肌病可能的致病基因和突变,仍然集中在五个桥粒基因,还有两个非桥粒基因TMEM43和PLN[7,18-21]。从表1可以发现,已报道的累及左心室致心律失常心肌病的非桥粒基因不多,整体上致病相关基因的纯合突变较多[18,19]。不同于传统ARVC最常见的致病基因是PKP2,目前报道的左心室受累的致心律失常心肌病最常见的致病相关基因突变是DSP基因。在53例年龄≤10岁的儿童病例中发现,携带DSP突变基因更容易累及左心室[22]。在一项134例散发病例中发现,与PKP2,DSG2,DSC2等突变基因携带者相比,DSP突变基因携带者更容易累积左心室[23]。从分子层面上来看,DSP基因中导致氨基酸链合成提前终止的突变更多,这类突变患者多表现为显著的左心室射血分数降低,影像学上有更大范围的左心室延迟增强,心电图表现为频发的侧壁T波倒置和右束支阻滞形室性心动过速。而携带DSP基因框移突变和无义突变的患者临床上多发生频发的短阵室性心动过速等室性心律失常[1]。综合分析推测DSP基因突变可导致其编码蛋白C末端的合成提前终止,无法与中间丝有效结合,从而与心肌结蛋白失去相互作用,由此导致严重的左心室病变[8,9]。
DSP基因突变的致病作用在小鼠动物模型中得到验证,通过建立心肌特异的DSP基因敲除小鼠模型(DSP-cKO)发现,DSP-cKO小鼠与对照组相比,其左、右心室和室间隔心肌被纤维脂肪组织替代,心脏明显扩大。MRI检查发现了小鼠左、右心室容量的改变。后期DSP-cKO小鼠出现心力衰竭表型并死亡,类似左右心室型致心律失常心肌病的病理改变[24]。
另外动物模型研究已经证实PLN基因突变参与了左心室病变的致病机制。在一个LDAC的西方人种家系中发现PLN基因c.40_42delAGA突变与疾病共分离。通过转基因小鼠过表达有c.40_42delAGA突变的PLN基因导致转基因小鼠过早死亡。 PLN参与维持心肌细胞钙离子的稳态平衡,该突变可能影响PLN蛋白功能进而导致对肌浆网重新摄取钙离子不可逆转的超抑制和左、右心室心肌纤维化,从而发生LDAC[20]。
然而基因突变并不是决定致心律失常心肌病临床表型的唯一因素。有研究分析了231例致心律失常心肌病患者的基因突变和临床表型,证实基因突变的异质性,基因修饰作用和环境因素都会对不同亚型的出现产生一定的影响[25]。有研究发现,与静息状态相比,运动中右心室压力升高170%,而左心室压力升高23%,激烈运动更容易导致右心室功能障碍[26]。基因突变对表型的影响还有剂量效应,如致心律失常心肌病的一种特殊类型Carvajal综合征,是DSP基因纯合突变疾病,临床表型同时累及左、右心室,纯合突变可能具有剂量效应导致双心室受累[27]。另外有研究发现,同时携带多个突变的患者与携带单一突变的患者相比,临床表现更加严重,如重度右心室扩张,更高可能性累及左心室,危及生命的心律失常事件,甚至是猝死[28]。
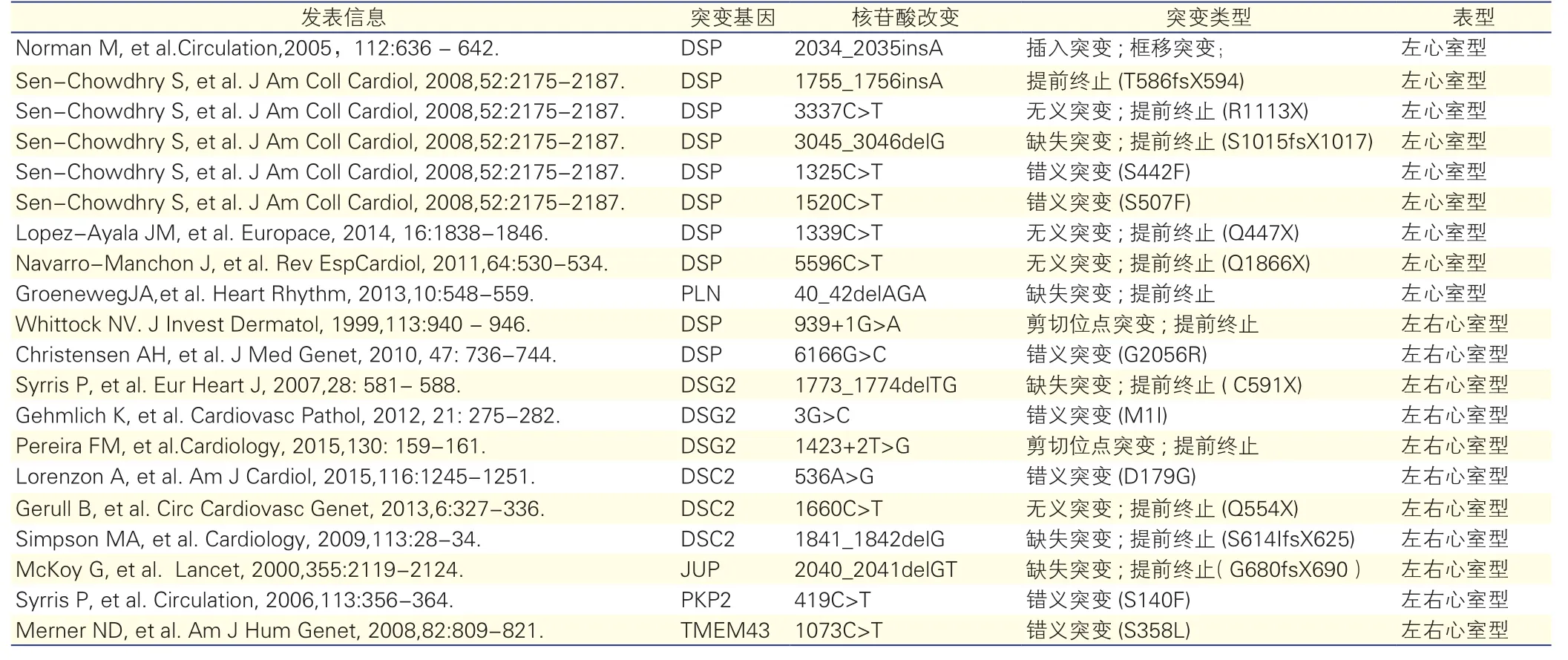
表1 累及左心室的致心律失常心肌病的相关基因突变
4 小结
总之,目前发现左心室受累的致心律失常心肌病比例并不低,临床表现多样,心律失常多表现为左心室起源的室性早搏或室性心动过速,心电图有左束支阻滞、电轴左偏、侧壁或下壁导联的epsilon波和T波倒置等,心脏超声可出现左心室结构和功能异常,右心室与左心室容量比<1。磁共振检查可发现左心室心外膜下或中层呈圆周状分布的延迟增强。左心室受累的致心律失常心肌病预后较差,易发生恶性室性心律失常导致心原性猝死。目前可考虑射频消融减少室性心律失常发作,并植入ICD预防心原性猝死。目前已报道的累及左心室致心律失常心肌病的致病基因多为桥粒基因,最常见的是DSP基因,多为纯合突变。但基因突变类型是否与致心律失常心肌病的类型有关目前尚不清楚。国内关于左心室受累的致心律失常心肌病的流行病学、临床预后和发病机制尚需要进一步研究。
[1] Sen-Chowdhry S, Syrris P, Ward D, et al. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation, 2007, 115: 1710-1720.
[2] Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace, 2011, 13: 1077-1109.
[3] Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res, 2015, 116: 1887-1906.
[4] Corrado D, Basso C, Thiene G, et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/ dysplasia: a multicenter study. J Am Coll Cardiol, 1997, 30: 1512-1520.
[5] Tavora F, Zhang M, Franco M, et al. Distribution of biventricular disease in arrhythmogenic cardiomyopathy: an autopsy study. Hum Pathol, 2012, 43: 592-596.
[6] De Pasquale CG, Heddle WF. Left sided arrhythmogenic ventricular dysplasia in siblings. Heart, 2001, 86: 128-130.
[7] Sen-Chowdhry S, Syrris P, Prasad SK, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol, 2008, 52: 2175-2187.
[8] Bauce B, Basso C, Rampazzo A, et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J, 2005, 26: 1666-1675.
[9] Sen-Chowdhry S, Syrris P, Mckenna WJ. Desmoplakin disease in arrhythmogenic right ventricular cardiomyopathy: early genotypephenotype studies. Eur Heart J, 2005, 26: 1582-1584.
[10] Lopez-Ayala JM, Gomez-Milanes I, Sanchez MJ, et al. Desmoplakin truncations and arrhythmogenic left ventricular cardiomyopathy: characterizing a phenotype. Europace, 2014, 16: 1838-1846.
[11] 黄洁, 郑哲, 胡盛寿, 等. 致心律失常性右心室心肌病累及左心室的临床病理分析. 中国循环杂志, 2007, 22: 278-281.
[12] McKenna WJ, Thiene G, Nava A, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Br Heart J, 1994, 71: 215-218.
[13] Te RA, Tandri H, Bluemke DA. Arrhythmogenic right ventricular cardiomyopathy (ARVC): cardiovascular magnetic resonance update. J Cardiovasc Magn Reson, 2014, 16: 50.
[14] 黄静涵, 孙兴国, 赵世华, 等. 比较心电图与超声心动图及磁共振成像诊断致心律失常性右心室心肌病的特征及诊断意义. 中国循环杂志, 2013, 28: 330-333.
[15] Protonotarios N, Tsatsopoulou A, Anastasakis A, et al. Genotypephenotype assessment in autosomal recessive arrhythmogenic right ventricular cardiomyopathy (Naxos disease) caused by a deletion in plakoglobin. J Am Coll Cardiol, 2001, 38: 1477-1484.
[16] Friedberg MK, Redington AN. Right versus left ventricular failure: differences, similarities, and interactions. Circulation, 2014, 129: 1033-1044.
[17] Calore M, Lorenzon A, De Bortoli M, et al. Arrhythmogenic cardiomyopathy: a disease of intercalated discs. Cell Tissue Res, 2015,360: 491-500.
[18] Christensen AH, Benn M, Bundgaard H, et al. Wide spectrum of desmosomal mutations in Danish patients with arrhythmogenic right ventricular cardiomyopathy. J Med Genet, 2010, 47: 736-744.
[19] Gerull B, Kirchner F, Chong JX, et al. Homozygous founder mutation in desmocollin-2 (DSC2) causes arrhythmogenic cardiomyopathy in the Hutterite population. Circ Cardiovasc Genet, 2013, 6: 327-336.
[20] Groeneweg JA, van der Zwaag PA, Jongbloed JD, et al. Leftdominant arrhythmogenic cardiomyopathy in a large family: associated desmosomal or nondesmosomal genotype?. Heart Rhythm, 2013, 10: 548-559.
[21] Syrris P, Ward D, Asimaki A, et al. Clinical expression of plakophilin-2 mutations in familial arrhythmogenic right ventricular cardiomyopathy. Circulation, 2006, 113: 356-364.
[22] Bauce B, Rampazzo A, Basso C, et al. Clinical phenotype and diagnosis of arrhythmogenic right ventricular cardiomyopathy in pediatric patients carrying desmosomal gene mutations. Heart Rhythm, 2011, 8: 1686-1695.
[23] Rigato I, Bauce B, Rampazzo A, et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet, 2013, 6: 533-542.
[24] Lyon RC, Mezzano V, Wright AT, et al. Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum Mol Genet, 2014, 23: 1134-1150.
[25] Sen-Chowdhry S, Syrris P, Pantazis A, et al. Mutational heterogeneity,modifier genes, and environmental influences contribute to phenotypic diversity of arrhythmogenic cardiomyopathy. Circ Cardiovasc Genet,2010, 3: 323-330.
[26] La Gerche A, Burns AT, Mooney DJ, et al. Exercise-induced right ventricular dysfunction and structural remodelling in endurance athletes. Eur Heart J, 2012, 33: 998-1006.
[27] Protonotarios N, Tsatsopoulou A. Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol, 2004, 13: 185-194.
[28] Fressart V, Duthoit G, Donal E, et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace, 2010, 12: 861-868.
2016-03-11)
(编辑:汪碧蓉)
100037 北京市,中国医学科学院 北京协和医学院 国家心血管病中心 阜外医院 心律失常中心
范思洋 硕士研究生 主要研究方向为心律失常 Email:18638035988@163.com 通讯作者:樊晓寒 Email:fanxiaohan@fuwaihospital.org
R541
A
1000-3614(2016)09-0931-04 doi:10.3969/j.issn.1000-3614.2016.09.025
