心肌细胞死亡在心力衰竭发病中的作用
2016-10-20马晓庆钟栩
马晓庆 钟栩
心肌细胞死亡在心力衰竭发病中的作用
马晓庆钟栩
心力衰竭是心脏不能提供足够的血液来满足机体代谢需要的临床综合征,被认为是所有心脏疾病的最终共同途径,也是心脏病患者死亡的主要原因。近十年的研究表明,在心力衰竭的发病及发展的机制中,心肌细胞死亡起着重要作用。本文综述了心肌细胞死亡的不同形式,讨论其分子机制及相互联系,为心力衰竭提供新的潜在的治疗方向。
细胞死亡;细胞凋亡;细胞坏死;心力衰竭
细胞死亡主要有凋亡(apoptosis)和坏死(necrosis)两种形式,其在正常的生物体生命活动中起着至关重要的作用,包括胚胎时期的发育和出生后机体的稳态调节[1],因此,许多疾病(如心血管疾病,糖尿病[2],癌症[3]等)发病机制与细胞死亡的增加或减少密切相关。本研究就细胞死亡在心力衰竭发病机制中的作用作一综述。
1 心肌细胞死亡的概述
细胞凋亡(apoptosis),是细胞的主动死亡,其过程受基因高度调控,细胞凋亡常散发在单个细胞,表现为细胞皱缩,包膜内陷、分裂成细胞膜封闭的“凋亡小体”,被吞噬细胞或周围细胞识别、吞噬。其过程不引起炎症反应,ATP仍保持在一定的水平[4]。细胞凋亡在多细胞生物的胚胎发生,发育、成熟中起重要作用。胚胎期间隔膜细胞的凋亡决定房、室间隔厚度[5]。成熟个体心肌细胞作为终末分化细胞,是否存在凋亡仍有争议。细胞坏死(necrosis)指在外界不良因素的刺激下,细胞病理性被动死亡,常波及成群细胞,表现为细胞膜通透性增加,细胞器及细胞肿胀,崩解死亡,其过程中,线粒体严重损伤,ATP生成显著降低甚至消失,引发明显炎症反应。细胞坏死历来被认为是不受调控的被动性死亡。1997年,Vercammen等研究发现,在肿瘤坏死因子-α(TNF-α)的诱导下,细胞可以同时出现凋亡和坏死的特征。近十年,越来越多的研究提示某些信号转导通路可以被细胞坏死与细胞凋亡共享,这种受细胞内信号严密调控的主动细胞坏死称为细胞程序性坏死(programmed cell necrosis,PCD)。自噬是细胞降解自身不需要的蛋白质和细胞器,有效控制细胞内蛋白质、细胞器质量,同时,参与细胞内物质能量循环利用的过程[6]。表现为内质网脱落的双层膜包裹蛋白聚合物和损伤的细胞器形成自噬小体,溶酶体分解其成分,同时提供细胞代谢能量。心肌细胞适当水平的自噬是一种生存机制,通过降解异常蛋白和衰老的细胞器,为细胞存活提供能量基础,同时还降解受损线粒体,阻断细胞凋亡[7]。过度自噬则会引起细胞损伤甚至死亡,称为细胞自噬(autophag)相关性细胞死亡,参与心血管疾病的发生和发展。
2 细胞死亡的途径
2.1外源性途径(死亡受体途径) 各种死亡配体结合同源受体触发细胞死亡。研究表明,相同的死亡配体根据下游事件的介导可以诱导细胞凋亡或坏死。配体和受体结合诱导形成死亡诱导信号复合物(DISC)和复合体I(complex I)[8]。死亡配体与死亡受体结合后,诱导胞内受体结构域构象发生变化,招募一个适配器蛋白(如Fas相关死亡域(FADD)),形成DISC,同时该适配器蛋白结合并激活上游Caspase8酶原,细胞凋亡。复合体I(complex I)由TNF受体相关因子2和5(TRAF 2 and 5),细胞凋亡蛋白的细胞抑制剂(IAP)1和2,受体相互作用蛋白1(RIP1)及肿瘤坏死因子受体(TNFR)组成[9]。在TNF-α的刺激下,复合体I解离TRAF2,TRADD,RIP1,其中RIP1去泛素化后,TRADD招募FADD与受体相互作用蛋白3(RIP3)形成复合体II(complex II)[10],其中的FADD结合并激活Caspase8前体,凋亡发生。实验研究表明,如果Caspase-8活性由病毒或癌蛋白抑制,凋亡途径受阻,细胞进入坏死途径,RIP1和RIP3相互交叉磷酸化,激活下游死亡信号[11]。
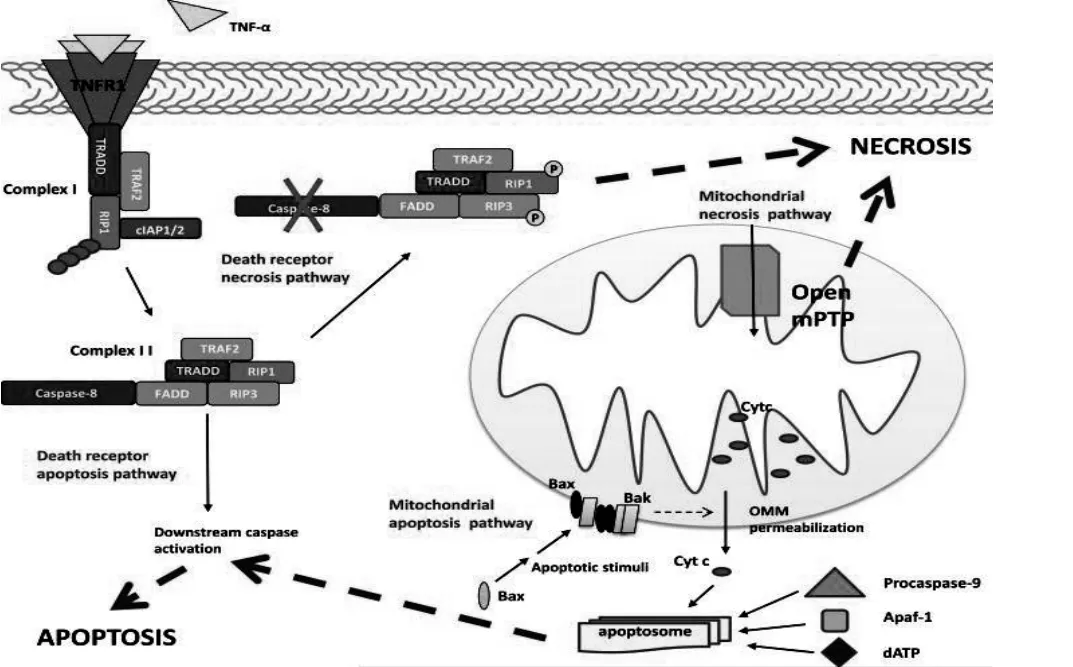
图1 细胞死亡途径(Arterioscler Thromb Vasc Biol.2012 Jul;32(7))
2.2内源性途径
2.2.1线粒体途径。通常被外界毒素、缺血、缺氧、DNA损伤等刺激激活。研究发现,线粒体介导细胞凋亡与坏死的机制区别在于前者导致线粒体外膜(OMM)通透性增加释放凋亡诱导介质,后者是线粒体通透性转换孔(mPTP)开放。大多数细胞死亡是内源性途径扩大外源性途径协同完成的。由抗凋亡蛋白(Bcl-2、Bcl-xL等)和促凋亡蛋白(Bax、Bak等)组成Bcl-2家族蛋白在线粒体凋亡通路的调控中起着重要作用。正常细胞胞质中的Bax蛋白受到“死亡刺激”后构象激活,迁移到线粒体外膜上,增加OMM通透性,释放细胞色素C,在ATP存在的条件下,细胞色素C联合凋亡蛋白活化因子(Apaf-1)激活Caspase-9前体,引起细胞凋亡[12]。当坏死刺激如Ca2+浓度过高,触发mPTP持续开放,导致线粒体膜跨膜电位(△Ψm)消失,ATP合成终止,此外,胞质中大量水涌入线粒体基质,线粒体肿胀、破裂,释放细胞色素C并活化Caspase[13]。
2.2.2内质网途径。内质网介导多种蛋白的合成,正确折叠,翻译后修饰以及钙离子稳态的维持。如当内质网Ca2+稳态发生变化,造成蛋白质合成折叠错误或未折叠蛋白增多,诱发内质网应激,若应激后激活相关转录级联,重建内质网的平衡状态,则为保护性机制,若过度应激则激活相关凋亡分子,细胞死亡。如错误折叠的蛋白激活RE1,进一步激活caspase12,细胞凋亡[14]。当Ca2+超载,通过内质网进入胞浆的同时增加线粒体内的Ca2+,细胞发生肿胀,坏死。进入胞浆内的Ca2+可以活化前凋亡蛋白Bad,促进细胞色素C释放,引起细胞的凋亡[15]。
2.3细胞自噬相关性细胞死亡目前对于自噬相关性细胞死亡精确机制尚未明确,但关于细胞自噬与细胞死亡之间蛋白质的联系有些已被确认,为进一步研究提供参考。Beclin-1蛋白是定位于内质网上含有与BH3-only类似结构域的蛋白,参与自噬体形成,促进细胞凋亡,其通常与Bcl-2形成复合物抑制细胞自噬。当线粒体损伤时,促细胞凋亡Bnip3(隶属于Bcl-2家族BH3-only亚家族)表达增多,与抗凋亡蛋白Bcl-2竞争性结合并激活Beclin-1蛋白,促进自噬体的形成。此外,研究表明Bcl-2-Beclin-1复合体中的Bcl-2可以被多个含有BH3-only的蛋白可以代替,促进细胞自噬[16]。
综上所述,细胞的死亡途径并非单一对应联系,而是不同途径上存在诸多交汇点。在死亡受体途径,Caspase8活性是细胞凋亡或坏死的关键点。Caspase8同时可激活促凋亡蛋白Bax,增加OMM通透性,释放细胞色素C,将死亡受体途径和线粒体途径联系起来。Ca2+超载时激活内质网RE1通路,活化caspase12,细胞凋亡,同时线粒体Ca2+升高,mPTP开放,细胞坏死,Ca2+对内质网及线粒体途径也起到连接作用。
3 细胞死亡与心力衰竭
3.1细胞凋亡与心力衰竭心衰患者心肌细胞凋亡率为0.08%~0.25%,是正常对照组(0.001%~0.01%)的10到100倍[17]。Caspase8转基因大鼠心肌细胞凋亡率0.023%(正常组的10倍)即可引起重度扩张性心肌病[18]。这些研究阐明了细胞凋亡诱导心力衰竭的充分性,其必要性通过抑制细胞凋亡可减轻心力衰竭的程度来表明。应用Caspase抑制剂可以观察到Caspase8转基因大鼠中心肌细胞凋亡率下降,心功能下降亦减轻。Vijayakumar等[19]发现奥美沙坦能抑制内质网应激引起的心肌细胞凋亡,改善自身免疫性心肌病动物的心功能。
3.2细胞坏死与心力衰竭钙超载的转基因小鼠模型中,心肌细胞过表达β2a亚基L型Ca2+通道,引起钙超载,mPTP开放,心肌坏死,心力衰竭[20]。在该模型中,过表达抗凋亡蛋白Bcl-2对抑制心衰无效,Cyclophilin D的缺失能减少心肌坏死。同样对ppif(编码亲环蛋白D)基因的敲除,可以改善阿霉素心肌病导致的心力衰竭。以此,我们推断细胞坏死可能与心力衰竭存在因果关系。
3.3自噬相关性细胞死亡与心力衰竭在心力衰竭既往的研究中表明,自噬相关性细胞死亡是心力衰竭中最常见的细胞死亡形式[21]。敲除Atg5基因的大鼠,自噬抑制,在压力超负荷状态下心室扩大,心力衰竭,表明自噬是心力衰竭的一种代偿性保护机制[22]。相反,Beclin-1+/-基因小鼠,过表达Beclin-1,自噬加强,但在压力超负荷条件下,病理性心肌重塑加重[23]。针对Atg5,Beclin-1+/-相互矛盾的研究结果尚无明确解释,在压力超负荷导致的心衰中,自噬对心肌的保护机制与促细胞死亡机制的转折点有待进一步研究。
4 结语
研究表明细胞死亡在心力衰竭的发病机制中起关键作用。细胞死亡信号转导通路的可调节性成为操控细胞死亡途径的作用靶点,为减少心肌细胞丢失,延缓心力衰竭的进程提供可能的治疗优势,但抑制慢性细胞死亡的潜在致癌作用也不能忽视。总之,心肌细胞的死亡干预研究仍处于起步阶段,在对心力衰竭中调控死亡的分子机制的进一步研究,将有可能为心衰的治疗策略,干预疾病的发生、发展起到突破性进展。
[1]Fuchs Y,Steller H.Programmed cell death in animal development and disease[J].Cell,2011,147(4):742-758.
[2]Gurzov EN,Eizirik DL.Bcl-2 proteins in diabetes:Mitochondrial pathways of beta-cell death and dysfunction[J].Trends Cell Biol,2011,21(7):424-431.
[3]Hanahan D,Weinberg RA.Hallmarks of cancer:The next generation[J].Cell,2011,144(5):646-674.
[4]Sun XM,Butterworth M,et al.Caspase activation inhibits proteasome function during apoptosis[J].Mol Cell,2004,14(1):81-93.
[5]霍霞,黄许行,朴英杰.胚胎发育与细胞凋亡[J].解剖科学进展,1996,2(3):212.
[6]Mizushima N,Levine B,Cuervo AM,et al.Autophagy fights disease through cellular self-digestion[J].Nature,2008,451(7182):1069-1075.
[7]Meyer G R,Martinet W.Autophagy in the cardiovascular system[J].Arch Biochem Biophys,2009,1793(9):1485-1495.
[8]Kischkel FC,Hellbardt S,Behrmann I,et al.Cytotoxicity-dependent apo-1(fas/cd95)-associated proteins form a death-inducing signaling complex(disc)with the receptor[J].EMBO J,1995,14(22):5579-5588
[9]Micheau O,Tschopp J.Induction of TNF receptor imediated apoptosis via two sequential signaling complexes[J].Cell,2003,114(2):181-190.
[10]Mitomi J,Christoffemon DE,Ng A,et al.Identification of a molecular signaling network that regulates a cellular necrotic cell deathpathway[J]. Cell,2008,135(7):1311-1323.
[11]Holler N,Zaru R,Micheau O,et al.Fas triggers an alternative,caspase-8-independent cell death pathway using the kinase rip as effector molecule[J].Nat Immunol,2000,1(6):489-495.
[12]Luo X,Budihardjo I,Zou H,et al.Bid,a Bcl2 interacting protein,mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors[J].Cell,1998,94(4):481-490.
[13]Baines CP,Kaiser RA,Purcell NH,et al.Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death[J]. Global Change Biology,2012,46(7033):1387-1401.
[14]ZhangK,Kaufman RJ.The unfolded protein response:a stress signaling pathway critical for health and disease[J].Neurology,2006,66(2 Suppl1):S102-109.
[15]LambHK,MeeC,XuW,et al.The affinity of a major Ca2+binding site on GRP78 is differentially enhanced by ADP and ATP[J].J Biol Chem,2006,281(13):8796-8805.
[16]Levine B,Sinha S,Kroemer G.Bcl-2 family members:Dual regulators of apoptosis and autophagy[J].Autophagy,2008,4(5):600-606.
[17]Guerra S,Leri A,Wang X,et al.Myocyte death in the failing human heart is gender dependent[J].Circ Res,1999,85(9):856-866.
[18]Wencker D,Chandra M,Ngnyen K,et al.A mechanistic role for cardiacmyocyte apoptosis in heart failure[J].Clin Invest,2003,111(10):1497.
[19]Vijayakumar Sukumaran,Kenichi Watanabe,Punniyakoti T,et al.Olmesartan,an AT1 Antagonist,Attenuates Oxidative Stress,Endoplasmic Reticulum Stress and Cardiac Inflammatory Mediators in Rats with Heart Failure Induced by Experimental Auto-immune Myocarditis[J].International Journal of Biological Science tertilrlfili clciece,2011,7(2):154-167.
[20]Nakayama H,Chen X,Baines CP,et al.Ca2+and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure[J].J Clin Invest,2007,117(9):2431-2444.
[21]Kostin S,Pool L,Elsasser A,et al.Myocytes die by multiple mechanisms in failing human hearts[J].Circ Res,2003,92(7):715-724.
[22]Nakai A,Yamaguchi O,Takeda T,et al.The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress[J]. Nat Med,2007,13(5):619-624.
[23]ZhuH,TannousP,JohnstoneJL,etal.Cardiacautophagyisamaladaptive responsetohemodynamicstress[J].JClinInvest,2007,117(5):1782-1793.
A
1004-2725(2016)04-0264-04
吴阶平医学基金会临床科研专项资助基金(项目编号:320.6750.11053)
730000甘肃 兰州,甘肃中医药大学中西医结合学院(马晓庆);730020甘肃 兰州,甘肃省第三人民医院心血管内科(马晓庆);730000甘肃 兰州,甘肃中医药大学附属医院心血管内科(钟栩)
马晓庆,E-mail:mxqing315@163.com
