常染色体显性遗传骨硬化症Ⅱ型氯离子第七通道蛋白基因突变1例检测
2016-08-11庞倩倩
庞倩倩,董 进
常染色体显性遗传骨硬化症Ⅱ型氯离子第七通道蛋白基因突变1例检测
庞倩倩,董进
山西医科大学第一医院(太原 030001)
关键词:遗传性骨病;骨硬化症;基因突变;氯离子第七通道蛋白;常染色体;检测
骨硬化症是一类以骨密度增高,破骨细胞吸收功能障碍为主要特点的遗传性骨病,根据其临床表现和致病基因的不同可分为常染色体显性遗传骨硬化症(autosomal dominant inheritance osteopetrosisADO),常染色体隐性遗传骨硬化症(autosomal recessive inheritance osteopetrosis,ARO)和罕见的X染色体连锁隐性遗传骨硬化症(X-linked osteopetrosis,XLO)。骨硬化症的临床表现具有广泛的异质性,如贫血、全血细胞减少、脓毒血症、继发性的肝脾肿大等。然而部分病人亦可无症状或症状较轻微,仅仅通过骨骼的影像学检查才可以发现。在影像学上该疾病通常表现为“中心性骨硬化”和“弥漫性骨硬化”两种类型,而这两种类型均由于骨脆性增加而引起的骨折风险率增加[1]。根据其临床表现不同,ADO又可分为:常染色体显性遗传骨硬化症良性Ⅰ型(ADO-Ⅰ),良性Ⅱ型(ADO-Ⅱ),良性Ⅲ型(ADO-Ⅲ)[1-2],其中ADO-Ⅱ最为常见。该型具有典型的ADO影像学特征,主要的并发症体现在骨骼系统,包括病理性骨折,脊柱侧凸等[3]。ADO-Ⅱ主要是由位于染色体16p13,编码氯离子第7通道蛋白(CLCN7)基因突变引起的[4]。目前国内罕有关于该疾病基因突变的报道,且尚未建立中国人群关于该疾病的基因突变谱,因此对骨硬化病人进行基因分析具有很重要的临床意义。
本研究拟通过对1例常染色体显性遗传的骨硬化症家系进行临床及基因学特征分析,探讨该疾病的致病机制。
1资料与方法
1.1研究对象先证者,男性,18岁。2015年因“骨硬化症”,就诊于我科门诊。病人第一胎,足月顺产,出生体重、身长、出牙时间均不详。智力未见落后。实验室检查结果:生化指标未见明显异常,全血细胞计数,血钙,血磷,血碱性磷酸酶,血肌酐,血iPTH水平均处于正常范围;影像学特征:头颅正侧位片示:颅骨骨皮质密度增厚,板障增厚,颅底骨质增厚硬化;胸腰椎正侧位相示:椎体上下终板骨密度增厚,中央密度相对较低,呈“三明治”样改变(见图1)。家族史中未有类似症状。病人及其父母均被告知研究目的,并签署知情同意书。
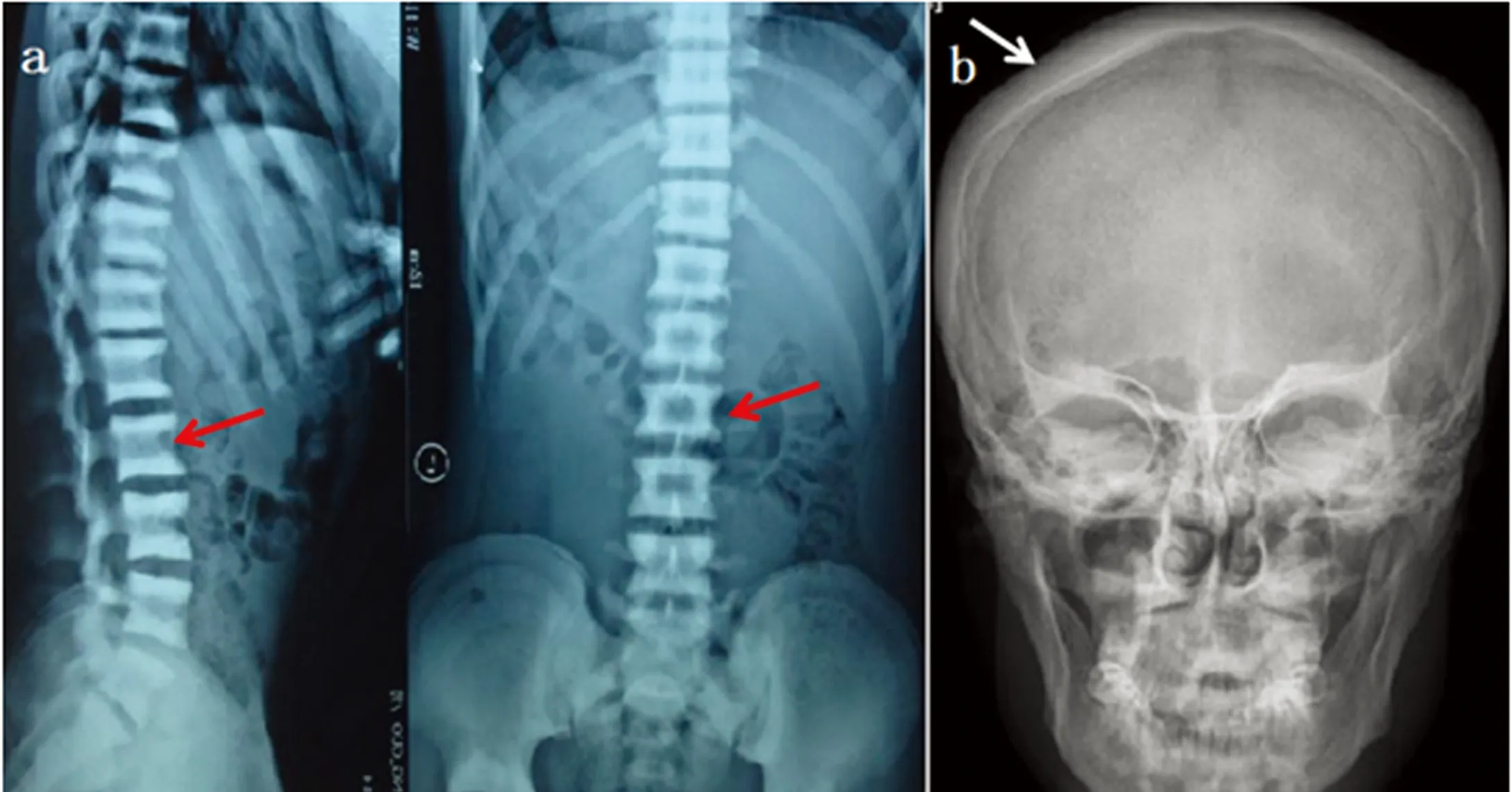
注:a.胸腰椎正侧位相:椎体上下终板骨密度增厚,中央密度相对较低,呈“三明治”样改变(红色箭头所示);b.头颅正位相:颅骨骨皮质密度增厚,板障增厚,颅底骨质增厚硬化(白色箭头所示)。
图1先证者影像学特征
1.2血标本的采集及DNA的制备在遵循知情同意原则的基础上,采集病人及其父母各2 mLEDTA抗凝血,用德国Qiagen全血DNA提取试剂盒提取病人及其家属的外周血DNA。
1.3致病基因外显子引物设计根据NCBI提供的人类基因组检索网站(http://www.ncbi.nlm.nih.gov/gene),对人类野生型CLCN7的基因组序列进行检索,使用专业生物学引物设计软件GeneRunner software对该基因的编码外显子及其交界区进行PCR引物设计,经UCSC验证,确保所有的引物都具有高度的特异性,扩增片段长度及退火温度见表1。
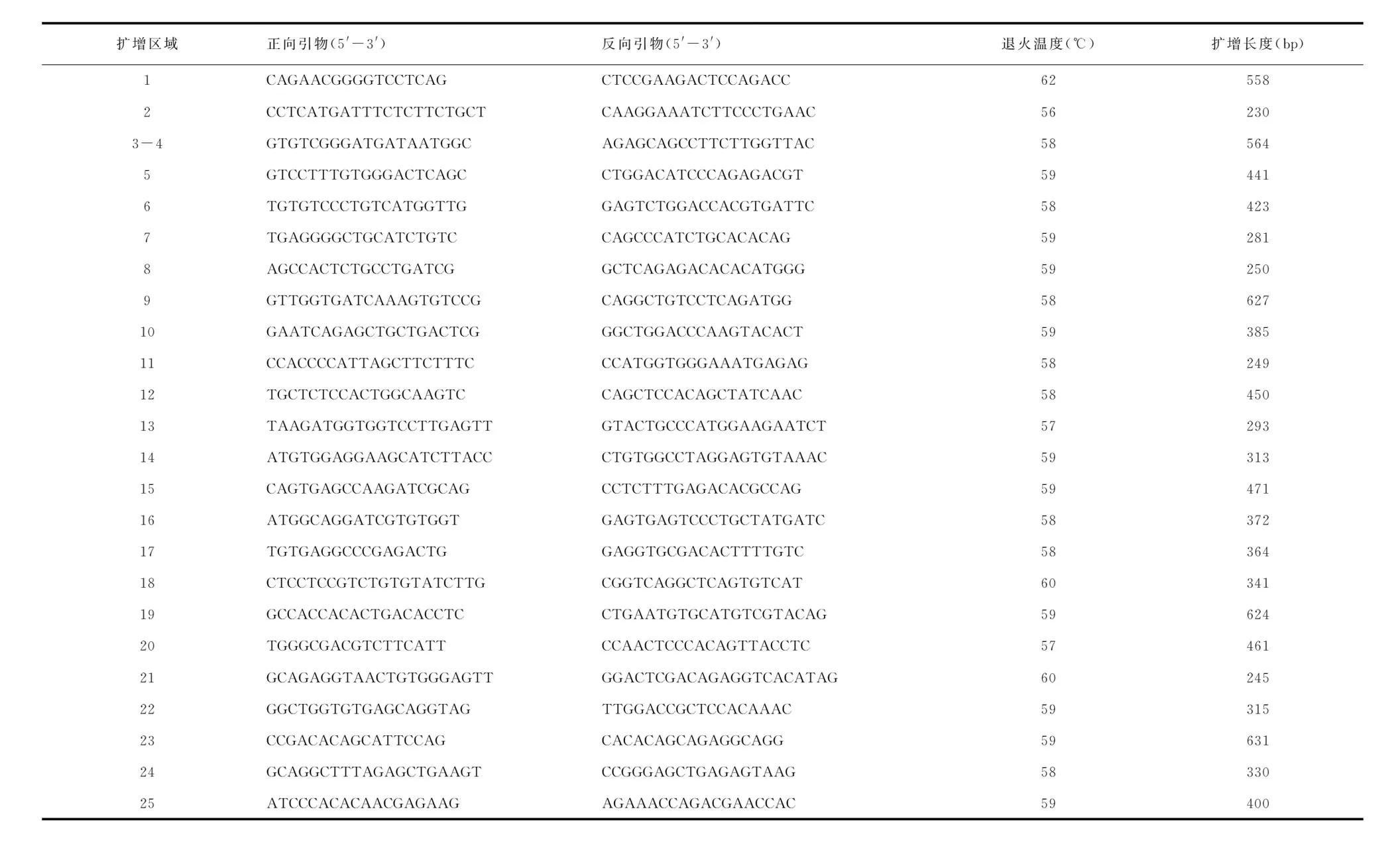
表1 CLCN7基因引物序列
1.4PCR扩增测序总反应体系30 μL:DNA模板2.6 μL,上下游引物各1.2 μL,2×Taq PCR 混合酶15 μL,ddH2O 10 μL;反应条件:预变性 95 ℃ 5 min,变性 94 ℃ 30 s,退火56 ℃ ~62 ℃ 30 s,延伸 72℃ 1 min,循环35轮,末次延伸 72℃ 10 min。
PCR产物测序:PCR产物送专业生物公司完成测序,使用美国A&B公司(Applied Biosystems,Foster City,CA)ABI3770 测序仪测序。
1.5DNA序列突变分析应用Chromas Version2.4软件分析测序结果,将测序图与正常的序列比对,然后应用UCSC和dbSNP138数据库分别与基因组DNA序列进行比对。最后应用Human Gene Mutation Database Professional 2014(HGMD 2014)数据库对突变体进行检索,确定其是否为新发突变。对所有测序反应均双向验证。
2结果
该家系图及测序结果见图2。先证者及其父母CLCN7基因突变检测结果示:先证者为第23外显子c.2299C>T(p.R767W)的杂合错义突变;其父母均非该突变的携带者,该突变为一自发突变。经查对2014HGMD突变数据库,证实该突变为一已知突变,在2001年由Cleiren首次报道[5]。
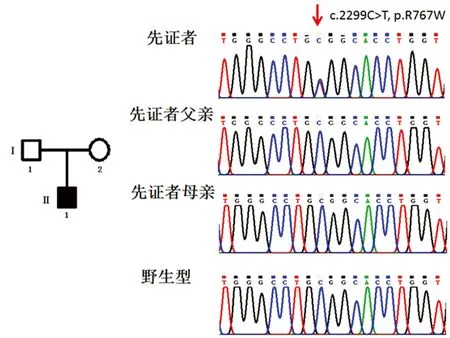
注:左边为家系图。黑色全黑方框为先证者(II1)。I1为先证者父亲,I2为先证者母亲,均非杂合子。右侧为CLCN7基因测序图。先证者为第23外显子为c.2299C>T(p.R767W)的杂合错义突变(箭头所示),其父母亲均非该突变携带者。
图2病人家系图及CLCN7基因测序结果
3讨论
ADO-Ⅱ又名“Albers-Schönberg病”[6],是一种罕见的代谢性骨病,该亚型的临床表现极度特异,且外显率不全。ADO-Ⅱ型具有典型的ADO影像学特征,如椎体上下终板骨密度增厚,中央密度相对较低“夹心椎”样表现及髂骨骨密度不均匀增高,出现浓淡相间的同心环状的“骨中骨”现象。该病主要的并发症体现在骨骼系统,包括病理性骨折、脊柱侧凸、髋骨骨关节炎和上颌骨多发骨髓炎[6]。
目前认为编码氯离子第7通道蛋白7基因突变是引起ADO-Ⅱ的主要原因[4]。CLCN7属于多通道膜蛋白,主要功能是通过电压-门控通道调节Cl-/H+交换。在破骨细胞中,CLCN7位于褶皱缘和溶酶体的隔室内,主要作用是将细胞外已通过离子交换进入细胞内的Cl-运输入破骨细胞的小囊泡,在小囊泡中与通过质子泵运输来的H+作用形成溶骨的酸性环境。破骨细胞吸收腔的低pH环境对于完成正常的骨吸收作用非常重要[7]。研究发现ADO-Ⅱ主要发病机制为CLCN7蛋白功能障碍,造成破骨细胞酸性环境形成受损,不足以发挥破骨细胞的骨吸收作用[4]。
本研究中先证者具有典型的骨硬化症影像学表现,对该先证者进行CLCN7基因检测,结果发现该病人CLCN7基因的第23外显子c.2299C>T(p.R767W)的杂合错义突变。该突变为一已知突变,但在国内罕有报道。Waguespack等[8]对62例ADO病人研究后发现,该R767W为较常见的一种。
本研究通过辅助检查和基因检测的方法,确证了1例中国人群罕有报道ADO-Ⅱ病人,进一步证实了CLCN7基因c.2299C>T (p.R767W)突变与该病人ADO-Ⅱ临床表现密切相关。
参考文献:
[1]Balemans W,Van Wesenbeeck L,VanHul W.A clinical and molecular overview of the human osteopetreses [J].Calcif Tissue Int,2005,77: 263-274.
[2]柯耀华,章振林.骨硬化症致病基因研究进展[J].中华骨质疏松和骨矿盐疾病杂志,2010,3(2): 1674-2591.
[3]Andrea Del Fattore,Alfredo Cappariello,Anna Teti.Genetics,pathogenesis and complications of osteopetrosis[J].Bone,2008,42: 19-29.
[4]Kasper D,Planells-Cases R,Fuhrmann JC,et al.Loss of the CLC-7 chloride channel leads to osteopetrosis in mice and man [J] Cell,2001,104: 205-215.
[5]Cleiren E,Benichou O,Van Hul E,et al.lbers-Schonberg disease (autosomal dominant osteopetrosis,type Ⅱ) results from mutations in the ClCN7 chloride channel gene[J].Hum Mol Genet,2001,10: 2861-2867.
[6]Zornitza Stark,Ravi Savarirayan.Osteopetrosis[J].Orphanet Journal of Rare Diseases,2009,4:5.
[7]Graves AR,Curran PK,Smith CL,et al.The Cl(/H)(+)antiporter ClC-7 is the primary chloride permeation pathway in lysosomes[J].Nature,2008,453: 788-792.
[8]Waguespack SG,Koller DL,White KE,et al.Chloride channel 7(ClCN7) gene mutations and autosomal dominant osteopetrosis,type Ⅱ[J].J Bone Miner Res,2003,18: 1513-1518.
(本文编辑王雅洁)
通讯作者:董进,E-mail: Sdyydj@medmail.com.cn
中图分类号:R596
文献标识码:C
doi:10.3969/j.issn.1672-1349.2016.13.048
文章编号:1672-1349(2016)13-1566-03
(收稿日期:2015-05-30)
