单相多铁性体CaMn7O12的理论研究与固相合成
2016-07-22张瑞浩代建清牛之慧程振宇王志翔昆明理工大学材料科学与工程学院昆明650093
张瑞浩 代建清 牛之慧 李 亚 程振宇 王志翔(昆明理工大学材料科学与工程学院,昆明 650093)
单相多铁性体CaMn7O12的理论研究与固相合成
张瑞浩代建清*牛之慧李亚程振宇王志翔
(昆明理工大学材料科学与工程学院,昆明650093)
摘要:在理论研究方面,采用基于密度泛函理论(DFT)的第一性原理方法对CaMn7O12的晶体结构进行了计算和表征,对螺旋桨型磁序的电子结构及相变机制进行了理论分析和研究;在实验方面以TG-DSC为指导,采用固相反应法制备了单相多铁性体CaMn7O12,并检测表征其磁、电学性能。磁学方面验证了CaMn7O12的2个磁相转变温度(TN1=90 K和TN2=45 K),测得其在10 K温度下存在磁滞回线(Mr=0.02 emu·g-1,Hc≈1 000 Oe);电学方面在室温条件下表征其在10 MHz频率时εr=280,tanδ=1.69。
关键词:CaMn7O12;第一性原理计算;固相反应法;电磁性能
近年来发展迅速的多铁性材料(multiferroics)是一类新型功能性材料,它的概念首先由瑞士学者Schmid[1]提出,此类材料同时具有2个或2个以上的铁性序(包括铁电序,(反)铁磁序,铁弹序等)[2-3]。根据铁电性的微观起源,单相多铁性体可分为2类[4]:第一类多铁性体(TypeⅠmultiferroics),铁电性和磁性有不同的起源。如BiFeO3[5-6],这类材料往往表现出较高的铁电临界温度和较大的极化强度[2-3],但缺点是铁电性与磁性耦合较弱,不利于实际应用;第二类多铁性体(TypeⅡmultiferroics),铁电性是由特定磁序引起的。这类多铁体由于本身特殊的磁致铁电性物理机制,其本征的磁电耦合效应较强,进而通过铁性的耦合协同作用产生一系列新的功能,从而受到了更为广泛的关注[7]。
国家自然科学基金(No.51162019,51462019)资助项目。
*通信联系人。E-mail:djqkust@sina.com
CaMn7O12作为一种新型的单相磁致多铁性体(TypeⅡmultiferroics),其铁电性是由结构中螺旋桨型磁序(proper-screw magnetic order)引起的,即材料的自发极化由磁结构引起[8]。具体来说,CaMn7O12共有2个磁相转变温度(TN1≈90 K,TN2≈48 K):在90 K时发生顺磁-反铁磁(PM-AFM)转变,在TN2<T<TN1温度区间内体系的磁结构是非线性长程螺旋磁有序的(AFMⅠ磁结构,也称为特殊的非共线反铁磁序);而当温度下降到TN2以下时,其表现反铁磁序(AFMⅡ磁结构),由于该结构比较复杂,故我们对其磁结构的研究以AFMⅠ结构为主[9]。CaMn7O12的磁致多铁性机制使得其自发铁电极化强度较大(P= 2870 μC·m-2)[10],铁电临界温度较高(TN1≈90 K)[11],且本征的磁电耦合效应更强。这种电极化和磁化通过内禀磁电作用的耦合,不仅为下一代多功能电子器件的设计提供了额外的自由度,而且大大拓宽了铁性材料的研究范围,对发展基于磁性-铁电集成效应的新型信息存储-处理磁电器件的应用技术提供了巨大的推动力,为磁性存储器件的快速读写和器件小型化的研究提供了充足的条件,特别是在微波领域、高压输电线路的电流测量、多功能电子设备(传感器、制动器、感应器、转换器等)方面提供了广泛的应用前景[12]。
本文采用基于密度泛函理论 (density functional theory,DFT)的第一性原理方法对CaMn7O12在440 K以下的三方晶系148号空间群R3晶体结构进行了计算和表征,并对其螺旋桨型磁序实验结构的态密度与相变机制进行了理论分析和研究。且在TGDSC热分析的指导下,确定CaMn7O12的烧结制度,采用固相反应法制备了化学成分准确、粒度分布均匀的CaMn7O12纯相,并对其各项性能作系统性的分析表征。
1 计算与实验方法
1.1计算方法
第一性原理计算采用基于密度泛函理论(DFT)的VASP软件包[13],并采用广义梯度近似(generalized gradient approximation,GGA)下的PBE泛函[14]。利用GGA+U的方法增强Mn的3d电子间的库仑排斥势,Ueff=2 eV。所有计算的平面波截断能都取500 eV,结构优化时高斯展宽宽度取0.05 eV,当Hellmann-Feynman力低于10 meV·nm-1时停止对离子位置和结构参数的弛豫。对于螺旋桨型磁序实验结构,布里渊区积分采用3×3×5 MP k点网格,计算态密度时采用7×7×9网格。参与计算的Ca,Mn 和 O的价电子组态分别为 3s23p64s2、3p63d54s2和2s22p4。
1.2实验方法
实验中,将99.99%(AR)的CaCO3和MnO2粉末根据物质的量之比nCa2+∶nMn4+=1∶7混合后,以w原料w磨球∶w无水乙醇=1∶1.5∶1的质量比置于球磨罐中,在150 r·min-1的转速下湿法球磨24 h,取出后在80℃的恒温干燥箱中烘干30 h,待粉体完全干燥后用玛瑙研钵研磨并过筛(400目筛网),以使粉体粒度分布均匀利于煅烧。在TG-DSC热分析的指导下制定烧结制度(在900℃初步煅烧,并在930℃完成烧结),先将处理好的粉体置入箱式电阻炉中以5℃·min-1的升温速率升至900℃并保温24 h,后以3℃·min-的降温速率降至室温,取出后再次进行研磨并过筛。将煅烧后的一部分粉体压片成型后(压片机压强为15 MPa)再次置入炉内,在930℃的空气环境下无压烧结48 h后 (5℃·min-1升温速率,3℃·min-降温速率至室温),就得到了纯相的CaMn7O12块材用以测量介电性能;其余粉末以相同条件焙烧后得到纯相的CaMn7O12粉体,进行物相分析和形貌表征,并检测其磁学性能。其中制备CaMn7O12纯相的化学反应方程式为:

实验采用德国Netzsch公司STA 449F3同步热分析仪(TG-DSC:空气气氛,测试温度25~1 000℃升温速率为10℃·min-1)对样品进行差热分析;采用日本理学D/max-2200型X射线衍射仪 (Cu靶Kα射线,波长0.150 4 nm,管电压30 KV,管电流20 mA,连续扫描速度10°·min-1,扫描范围为10°~70°分析样品成相情况;采用Tescan VEGA3 SBH型扫描电子显微镜(30 kV时分辨率3.0 nm,加速电压为0.2~30 kV)表征样品形貌;采用SQUID-VSM(测量温区5~150 K,升温速率为5 K·min-1)检测样品磁学性能;采用Agilent 4294A精密阻抗分析仪(impedanc analyzer:测试频率40 Hz~110 MHz,频率分辨率为1 mHz)检测样品的介电性能。
2 计算结果与分析
2.1CaMn7O12的晶体结构
随着温度的变化,CaMn7O12发生如下结构相变,即在440 K左右时由高对称的立方晶系204号空间群Im晶格结构(a=0.735 3 nm)转变为低对称的三方晶系148号空间群晶格结构 (a=1.044 1 nm,c=0.634 3 nm)[11,15],而此结构也是CaMn7O12的常态结构,故本文以R晶格结构为主要研究对象。如图1 (a),当温度小于440 K时,CaMn7O12属于AC3B4O12型结构体系 (AC3B4O12这一结构起源于钙钛矿家族的经典结构ABO3型[16],由于晶体结构畸变等原因致使原来的A位置分成了A和C两种位置),晶体结构内共有6种离子:Ca2+,Mn13+,Mn23+,Mn34+,O12-和O22-,其中Ca2+和Mn13+分别以1∶3的比例占据晶格结构的A位置和C位置,Mn23+和Mn34+分别以3∶1的比例占据晶格结构的B位置,O12-与O22-离子分别以1∶1的比例占据晶格结构的O位置[15]。此外,Mn1与周围的2个O1和2个O2离子形成Mn1O4四面体,Mn2与周围的2个O2和4个O1离子形成Mn2O6轴向压缩氧八面体,Mn3与周围的6个O2离子形成Mn3O6正的氧八面体,3种磁性锰离子的自旋方向都在ab平面内沿着c轴呈螺旋手征排列。
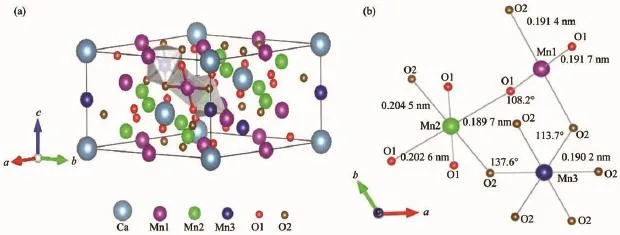
图1 (a)CaMn7O12的R3晶体结构;(b)ab平面内Mn1O4四面体和Mn2O6、Mn3O6八面体Fig.1 (a)R3 crystal structure of CaMn7O12;(b)Mn1O4tetrahedron and Mn2O6,Mn3O6octahedron in the ab plane
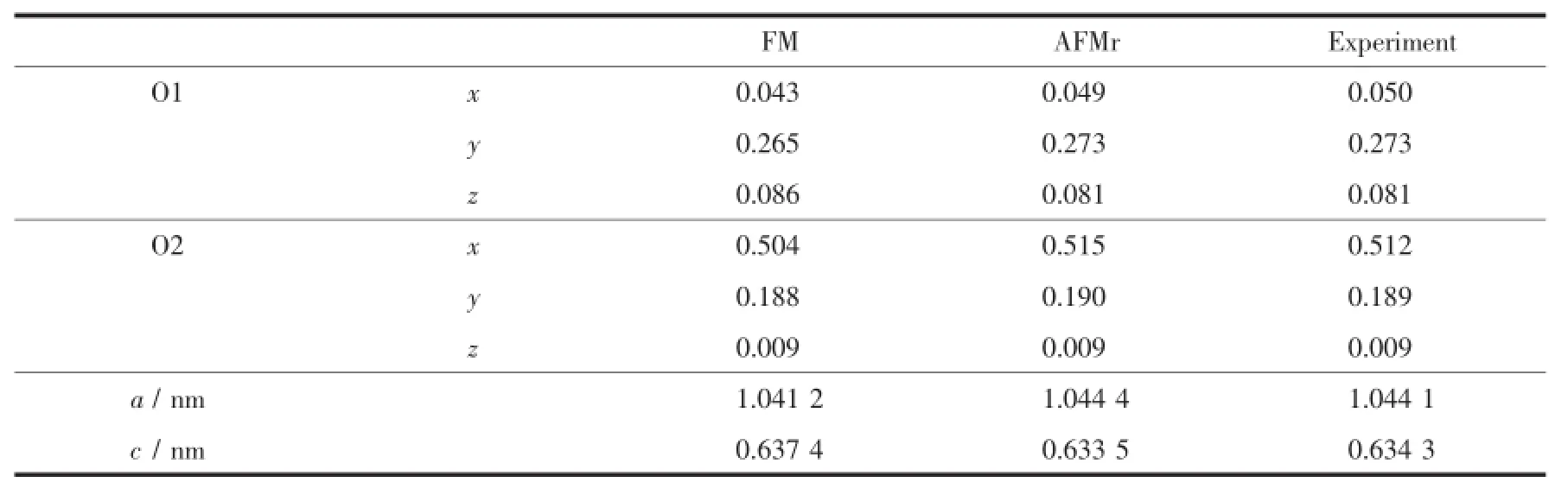
表1 FM、AFMr弛豫后的晶格参数和原子位置及其实验数据[17]Table 1 Lattice parameter and atom site of FM and AFM relaxation and the experimental data[17]
为在原有基础上进一步探究CaMn7O12在148号空间群下的R3晶格结构,我们采用第一性原理计算方法研究其AFMI磁结构 (在温度区间TN2<T<TN1时的非线性长程螺旋磁有序结构,其包含2种磁序,一种是铁磁序,另一种是调制磁有序[18])的Wyckoff位置,并推广得到晶格结构各离子的Wyckoff坐标。为选定AFMI结构的计算参考结构,我们分别对铁磁序FM和亚铁磁序AFMr两种线性磁序进行结构优化,找到它们的基态稳定结构后再与AFMI实验结构对比,得到物理性质与真实结构最接近的一种线性磁序。由于2种结构中都有3d电子,考虑到3d电子的强关联效应,我们采用对3d电子加U(在位库仑排斥势)的方法来增强电子间的库仑排斥作用(哈伯德参数U=3 eV,交换相互作用J=1 eV,Ueff=2 eV)。表1列出了弛豫后的晶体结构参数和原子位置,由表中数据可知,不论是O1、O2的原子位置还是晶格参数,亚铁磁序AFMr结构都比铁磁序FM结构更接近实验结构AFMI数据[17],因此本文选择亚铁磁序AFMr结构作为实验结构AFMI的参考结构。
在对亚铁磁序AFMr进行结构弛豫之后,推知CaMn7O12的R3晶格结构中Mn离子与周围O离子的具体位置关系。如图1(b),Mn1与周围的2个O1 和2个O2离子形成Mn1O4四面体,Mn-O键长分别为0.191 4和0.191 7 nm;Mn2与4个O1和2个O2离子形成Mn2O6轴向压缩氧八面体,Mn-O键长分别为 0.189 7、0.202 6、0.204 5 nm;Mn3与 6个O2离子形成Mn3O6正的氧八面体,Mn-O键长为0.190 2 nm。晶体结构中键角分别为:∠Mn1-OMn2=108.2°,∠Mn1-O-Mn3=113.7°,∠Mn2-O-Mn3= 137.6°,且该数值与之前LU等的晶体结构计算结果[19](计算值分别为108.1°、113.3°、137.6°)非常吻合。由此可知,采用GGA+U增强Mn的3d电子间库仑排斥势的方法得到的计算结果准确,且与图1 (a)显示的晶格结构中各离子位置吻合度较高,并补充完善了实验结果[17]所欠缺的Mn离子与周围O离子的具体Wyckoff位置关系,对于今后单相多铁性体CaMn7O12的研究具有一定参考价值。
2.2电子结构与相变机制
在对CaMn7O12的R3晶格结构进行表征分析后,我们对AFMI螺旋桨型磁序结构的态密度分布作了进一步研究。图2为AFMI磁结构的总态密度(total DOS)以及 O2p、Mn3d的分波态密度(partial DOS)分布图,横坐标为系统总能与费米能的差值,纵坐标为态密度。由图2(a)总态密度可看出,在费米能级附近没有电子占据,表明CaMn7O12为绝缘体,其中带隙的大小为0.43 eV,与之前研究的计算结果0.45 eV[19]非常接近。对图2(a)进一步分析,发现在较大能量范围内 (-7.50 eV至3.23 eV),Mn离子以及O离子对价带顶和导带底都有贡献,且Mn离子的贡献较大;O离子的电子占据状态与总的电子占据状态相似,在费米面附近相邻的2个峰之间有比较窄的零占据态,说明该结构有窄带效应,与Zhang等之前的实验结果吻合[11]。而在费米面以上未被电子占据的空带主要集中在2 eV附近,并且被局域在约3 eV的能量范围内,由图可知Mn3d轨道电子为其主要来源。图2(b)中O1p与O2p的态密度大小基本一致,而图2(c)中Mn1d、Mn2d和Mn3d的态密度大小不尽相同,Mn1、Mn2离子的态密度明显大于Mn3离子,即Mn1、Mn2离子对整体态密度的贡献更大。且由图2(b)和(c)可以看出,O离子及Mn离子的态密度重心作用区间主要集中在-7.50~-2.50 eV、-0.90~-0.20 eV、0.23~3.23 eV能量范围内,且二者态密度图中均有明显的杂化峰出现,表明CaMn7O12的AFMI螺旋桨型磁序结构具有较强的p-d轨道杂化现象,该结论与之前研究结果一致[19]。
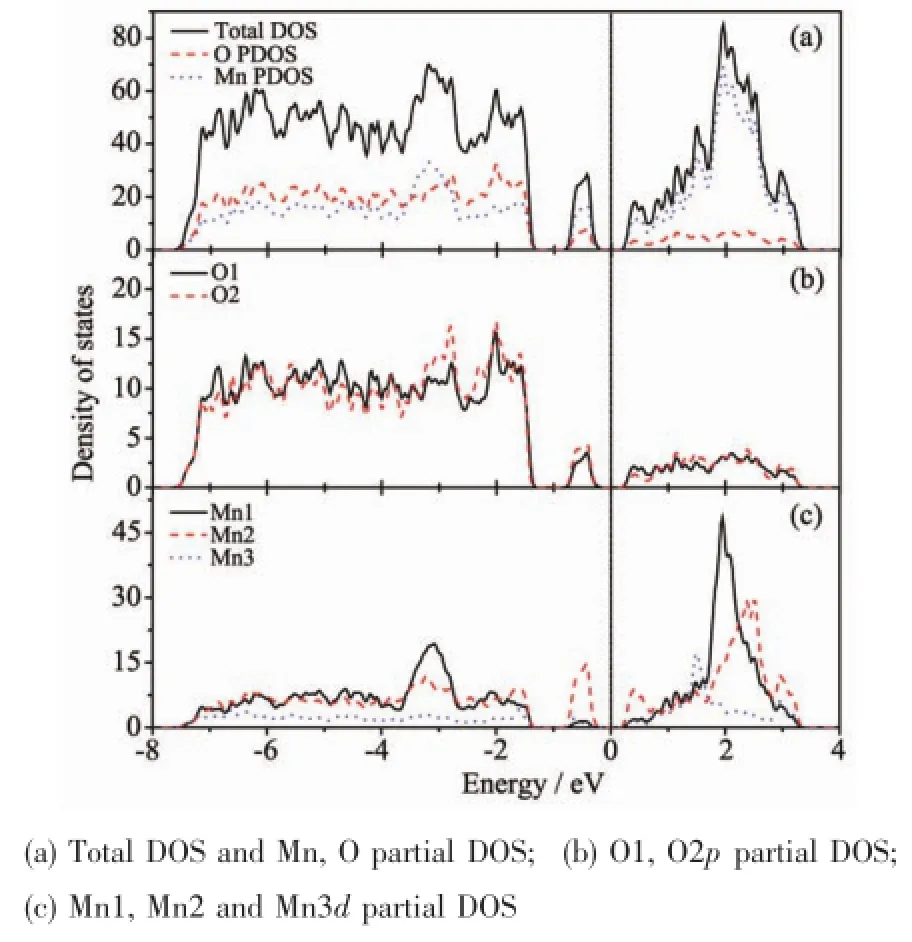
图2 CaMn7O12的AFMI结构Fig.2 AFMI structure of CaMn7O12
CaMn7O12磁相转变发生在 TN1≈90 K和TN2≈48 K 2个温度点附近,即由90 K以上的顺磁结构转变为90~48 K间的AFMI非线性长程螺旋磁序结构,继而到48 K以下复杂的AFMII反铁磁序结构[10-11];而与此同时,我们的计算[20]表明其本质的晶体结构也发生了转变 (但本文在计算方面仍采取以AFMI磁结构为参考结构推算R3晶格结构的方法[17,19]),即由90 K以上(440~90 K)的三方晶系148号空间群R3晶格结构转变为146号空间群R3晶体结构,继而到48 K以下的143号空间群P3晶体结构。也就是说在晶体结构转变过程中,R3结构为转变的基态,P3结构为转变的终态,R3结构是该转变过程的中间相态,其中第一阶段相变 (R3到R3结构)由螺旋桨型磁序下的磁诱导力引起,而第二阶段(R3到P3结构)的相变机制是在螺旋磁序结构的约束下,布里渊区边界处的软模推动了CaMn7O12由中间相态R3到基态结构P3的转变[21]。上述2次结构相变(由TN1以上到TN2以下)分别表现二级和一级相变特征,这也与实验中[10]CaMn7O12在TN1和TN2两个温度点所表现的磁相转变特征相吻合。
3 实验结果与讨论
3.1单相CaMn7O12的合成
在空气气氛中对前驱物粉体做热重及差示扫描量热(TG-DSC)分析,测试温度为25~1 000℃,升温速率为10℃·min-1。测试结果如图3,DSC曲线在100℃左右有一微小吸热峰,伴随TG曲线有不明显失重,分析为失去物理吸附水和游离水所致;随后DSC曲线在550、720、900℃出现连续的吸热峰,对应的TG曲线有3个失重台阶,共失重24%左右,可以判断样品在此过程中有结构水的散失和氧化还原反应的发生,并释放出CO2和O2,并在900℃附近发生结晶,从而导致了较大的热失重;在930℃时DSC曲线出现最后一个吸热峰,并伴随轻微失重,分析为CaMn7O12反应完成并完全结晶。观察整个TG-DSC曲线可知CaMn7O12晶体形成是缓慢渐进的,总反应过程失重在24%左右,与理论值23%非常吻合。分析其在900℃附近初结晶并在930℃完全结晶,故本文中实验部分据此制订了完善的烧结制度,并最终制得CaMn7O12纯相。

图3 CaMn7O12前驱体TG-DSC曲线Fig.3 TG-DSC curves of CaMn7O12precursor
观察由上述实验条件焙烧后得到粉体的衍射图(图4),所有衍射峰均可按照CaMn7O12的标准图谱(PDF:026-1114;三方晶系148号空间群R3晶体结构)进行指标化,且图中无其它相杂峰,表明实验得到了高纯度的CaMn7O12。
图5表征了CaMn7O12纯相粉体的SEM形貌,图中显示粉体颗粒呈长柱状均匀分布,且平均尺寸较小,粒度在0.5 μm左右。在一些结晶颗粒表面可以看到平整的晶面和明显的棱角,表明利用上述固相反应法制备得到质量优异的CaMn7O12粉体。

图4 CaMn7O12的X射线衍射图(a)和PDF卡片(b)Fig.4 X-ray diffraction pattern(a)and PDF card(b)of CaMn7O12

图5 CaMn7O12粉体SEM图Fig.5 SEM image of CaMn7O12powder
3.2CaMn7O12的性能表征
图6(a)显示了CaMn7O12在零场下冷却(ZFC)与外加磁场(H=500 Oe)下冷却(FC)的磁化率随温度(T= 5~150 K)变化的关系曲线。由图可知,其FC和ZFC曲线在T<45 K部分时有明显的分离,表明其在TN2=45 K发生磁相转变(反铁磁序称为AFMII)。这与较早文献中的实验结果TN2≈48 K[11]相吻合,并与Sannigrahi等在之前研究中[20]的发现相符:当温度为45 K时,CaMn7O12发生第一序磁相转变 (the first order nature of the transition)且基态是磁亚稳态。图6(a)插图表明在较高温度区间时(90~150 K),磁化率随温度的变化符合居里-外斯定律(Curie-Weiss law:χ=Cp/(T-θp),CaMn7O12此时表现良好的顺磁性(PM)。拟合曲线后可得居里-外斯温度θp=-53 K,而负值的θp标志着低温下存在反铁磁相互作用。Curie-Weiss现象和负的θp值也进一步证实CaMn7O12发生顺磁-反铁磁(PM-AFM)转变的相变温度为TN1=90 K(当温度在TN2<T<TN1之间时,体系表现非线性长程螺旋磁有序结构,磁结构为AFMI[15,18])。
本文进一步表征了CaMn7O12在低温下的磁滞回线。如图6(b)所示,在T<TN2温度下(T=10 K),CaMn7O12显示类铁磁性磁滞回线,这与Zhang等的报道一致[11],实验表明该温度范围下的AFMII反铁磁序在同一晶体学方向存在调制现象[10],此时形成的弱磁滞回线应该与此调制现象密切相关,但由于其具体磁结构太过复杂,其中的微观机理至今尚不明确;在T>TN1(T=100 K)时,由于物质表现顺磁性而测得其显示无矫顽力的线性M-H顺磁性回线。而在TN2<T<TN1温度范围内,中子衍射实验[10]已经确定CaMn7O12具有特殊的非共线反铁磁序,即AFMI非线性长程螺旋磁序。该磁序在外加磁场的作用下,由于产生了自旋沿磁场方向的倾斜 (spin canting),会出现宏观的磁化强度(比较微弱),但当外加磁场为零时,其自旋倾斜导致的宏观磁化能量不足以克服由温度条件所产生的热扰动能量,使得其剩余磁化为零而呈现顺磁行为[20]。总体来说,由于AFMI非共线结构具有反铁磁特征,使其在无外加磁场时不能产生净的自发磁化强度,这点与普通的共线反铁磁体(如NiO、MnO等)一致[21],因此CaMn7O12在70 K温度时同样表现宏观的顺磁性特性,即线性的M-H关系。图6(b)插图为H=0附近各回线的放大图,主要表征了CaMn7O12在T=10 K的温度情况下,剩余磁化强度Mr=0.02 emu·g-1,且矫顽力Hc≈1 000 Oe,相比之前溶胶-凝胶法(~450 Oe)[11]有所提升。
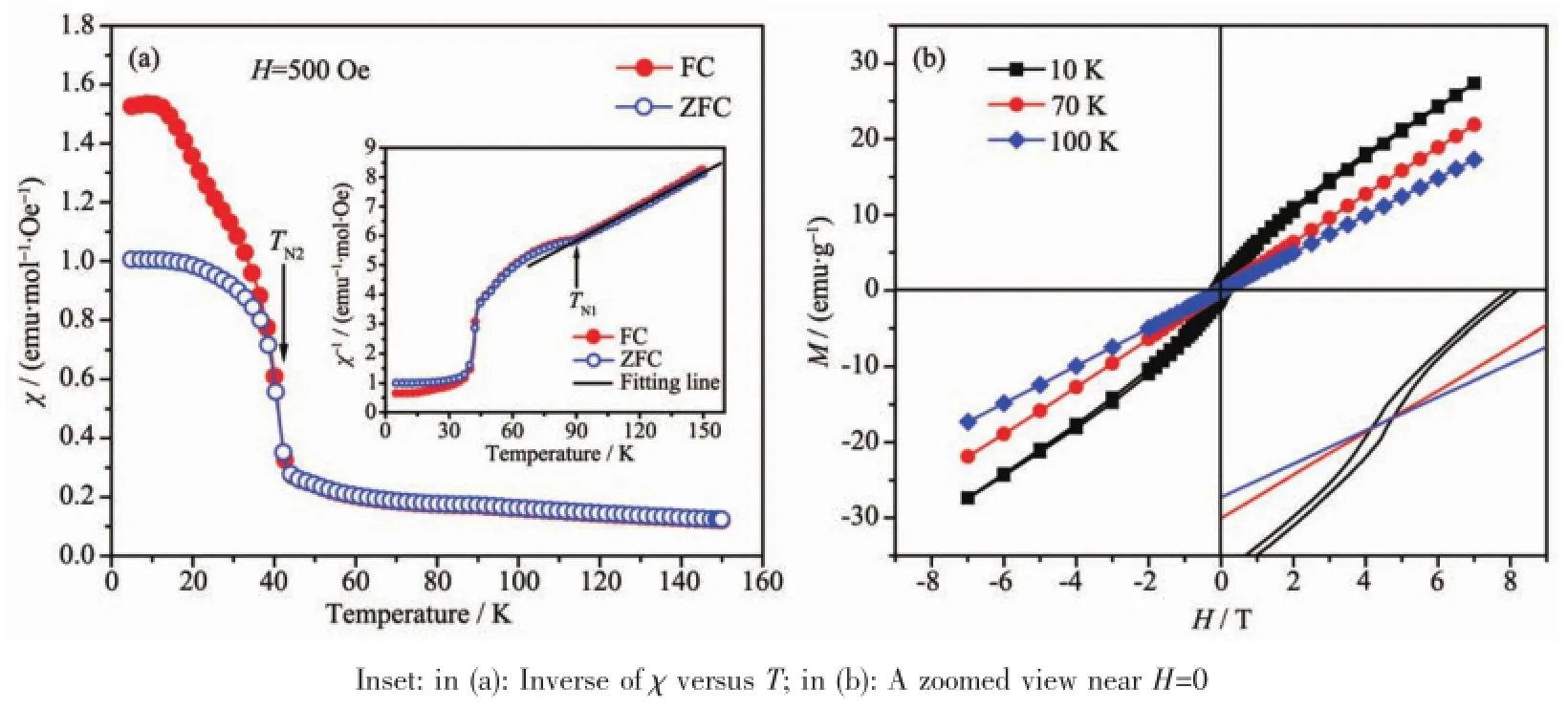
图6 CaMn7O12在零场和500 Oe场下磁化率随温度的变化曲线 (a)和不同温度下的磁滞回线(b)Fig.6 Temperature dependence of zero-field-cooled(ZFC)and field-cooled(FC)magnetic susceptibility(χ)measured in a field of 500 Oe(a)and magnetic hysteresis loops at different temperature(b)for CaMn7O12
在对CaMn7O12磁学性能进行表征研究后,为补充之前文献对其介电性能研究的不足之处,本文检测了CaMn7O12在室温条件下介电损耗和介电常数随频率的变化曲线,如图7。由图可知介电常数ε曲线和介电损耗tanδ曲线的变化规律基本一致:二者在初期随频率的增大迅速减小,在10 MHz频率附近趋于缓和后随频率缓慢减小。首先分析介电常数εr的变化:在频率增大初期,电介质中主要具有位移极化和松弛极化2种极化方式。其中松弛极化的产生机理与质点的热运动有关,由于质点需克服一定的势垒才能移动,因此这种极化建立的时间较长并且消耗能量。而随着频率的逐渐增大,用时较长的松弛极化来不及建立,介电常数仅由瞬时性的位移极化决定(相比松弛极化,位移式极化形成时间极短,外电场取消后能立即恢复到原来的状态,基本上不消耗能量,是电介质的主要极化方式),因此如图7中所示,介电常数值由于缺少松弛极化的贡献而迅速减小,并在位移极化的主导作用下(10 MHz以上)缓慢减小。而对于介电损耗tanδ曲线:电介质损耗主要有漏导损耗、极化损耗和共振吸收损耗3种。其中漏导损耗是由于电介质中的带点质点在外电场作用下运动而产生一定的电导,造成能量的损失。漏导损耗贯穿电介质损耗过程的始终,并在频率增大的初期,与松弛极化损耗(松弛极化的松弛时间较长,在电场作用下电偶极矩的取向跟不上电场变化而产生介电损耗)一同起主要作用。当外电场频率较高时,由于松弛极化来不及建立而无法产生极化损耗(位移极化无损耗),故在10 MHz以后介电损耗曲线在仅有漏导损耗的作用下趋于平稳(由于实验测试频率尚未达到CaMn7O12晶体发生共振吸收损耗所要求的频率最小值,故图7显示样品未发生共振吸收损耗)。综合上述变化规律,物质的2种介电曲线在10 MHz附近具有明显的转变特征,所以我们着重考察样品在该频率附近(见图7插图)的介电性能,由图可知,10 MHz时CaMn7O12的介电常数值εr=280,介电损耗值tanδ=1.69。

图7 CaMn7O12在室温下不同频率时的介电常数和介电损耗Fig.7 Dielectric constant and dielectric loss of CaMn7O12as function of frequency
4 结论
本文分别从理论计算研究和化学合成表征两方面对单相磁电多铁性体CaMn7O12进行了较为系统的研究和分析,表明计算与实验结果间存在相互吻合关系,并可在一定程度上互相补充。采用基于密度泛函理论的第一性原理方法对 CaMn7O12在440 K以下的三方晶系148号空间群R3晶体结构进行了计算表征,对实验结构螺旋磁序AFMI的电子结构和相变机制进行了分析研究。以此为参考,在TG-DSC热分析的指导下,确定了CaMn7O12的烧结制度,采用固相反应法制备了化学成分准确、颗粒粒径细小、形貌结构完整且分布均匀的CaMn7O12纯相,系统地表征了它的磁、电学性能。磁学检测结果显示其磁相转变温度分别为TN1=90 K、TN2=45 K,与之前研究非常吻合,而在10 K温度下的磁滞回线表明其剩余磁化强度Mr=0.02 emu·g-1,且矫顽力高达Hc≈1 000 Oe;电学性能测量表明,室温下CaMn7O12在频率为10 MHz时介电常数值εr=280,介电损耗值tanδ=1.69。
参考文献:
[1]Schmid H.Ferroelectrics,1994,162:317-338
[2]HE Hong-Cai(何泓材),LIN Yuan-Hua(林元华),NAN Ce-Wen(南策文).Chinese Sci.Bull.(科学通报),2008,10:1136-1148
[3]Hill N A.J.Phys.Chem.B,2000,104(49):6694-6709
[4]DONG Shuai(董帅),XIANG Hong-Jun(向红军).Physics(物理),2014,43(3):173-182
[5]QIU Zhong-Cheng(邱忠诚),ZHOU Jian-Ping(周剑平),ZHU Gang-Qiang(朱刚强),et al.Chinese J.Inorg.Chem.(无机化学学报),2009,25(4):751-755
[6]WU Zi-Wei(吴子伟),LÜ Xiao-Meng(吕晓萌),SHEN Jia-Yu(沈佳宇),et al.Chinese J.Inorg.Chem.(无机化学学报),2014,30(3):492-498
[7]CHI Zhen-Hua(迟振华),JI Chang-Qing(靳常青).Prog.Phys.(物理学进展),2007,02:225-238
[8]Przeniosło R,Wardecki D,Sławiński W,et al.Physica B,2013,428(1):27-29
[9]Zhang J T,Lu X M,Zhou J,et al.Phys.Rev.B,2013,87: 075127
[10]Johnson R D,Chapon L C,Khalyavin D D,et al.Phys.Rev. Lett.,2012,108(6):067201
[11]Zhang G Q,Dong S,Yan Z B,et al.Phys.Rev.B,2011,84: 174413
[12]Dong S,Liu J M.Mod.Phys.Lett.B,2012,26(9):1230004
[13]Gonze X,Lee C.Phys.Rev.B,1997,55:10355
[14]Perdew J P,Ruzsinszky A,Csonka G I,et al.Phys.Rev. Lett.,2008,100(13):136406
[15]Przeniosło R,Sosnowska I,Suard E,et al.J.Phys.:Condens. Mater.,2002,14(23):5747-5753
[16]Vasil′ev A N,Volkova O S.Low Temp.Phys.,2007,33(11): 895-914
[17]Resta R.Rev.Mod.Phys.,1994,66(3):899-915
[18]Sánchez-Andújar M,Yáñez-Vilar S,Biskup N,et al.J.Magn. Magn.Mater.,2009,321:1739-1742
[19]Lu X Z,Whangbo M H,Dong S,et al.Phys.Rev.Lett,2012,108(18):187204
[20]Dai J Q,Zhang H,Song Y M.New J.Phys.,2015,17: 113038
[21]Dai J Q,Zhang H,Song Y M.J.Magn.Magn.Mater.,2015,396:135-139
[22]Sannigrahi J,Chattopadhyay S,Dutta D,et al.J.Phys.: Condens.Mater.,2013,25:246001-246008
中图分类号:O641;O482
文献标识码:A
文章编号:1001-4861(2016)05-0762-07
DOI:10.11862/CJIC.2016.103
收稿日期:2015-11-06。收修改稿日期:2016-03-21。
Theoretical Calculations and Solid-Phase Synthesis of the Single Phase Multiferroic CaMn7O12
ZHANG Rui-HaoDAI Jian-Qing*NIU Zhi-HuiLI YaCHENG Zhen-YuWANG Zhi-Xiang
(Faculty of Materials and Metallurgical Engineering,Kunming University of Science and Technology,Kunming 650093,China)
Abstract:On theoretical aspect,The R3 crystal structure and the proper-screw magnetic order(the electronic structure and the transformation mechanisms)of CaMn7O12were calculated by using the first principles calculation based on the density function theory.The purity-phase CaMn7O12was prepared by solid reaction process according to the TG-DSC analysis in the experiment,which was known as a new single-phase multiferroic material,and its magnetic and electric properties were characterized successfully.Two magnetic phase transition temperature(TN1=90 K and TN2=45 K)and a hysteresis loop at 10 K(Mr=0.02 emu·g-1and Hc≈1 000 Oe)are measured by the magnetism performance testing.The electrical performance testing indicates that CaMn7O12has the dielectric constant εr=280 and the dielectric loss tanδ=1.69 at room temperature(at 10 MHz).
Keywords:CaMn7O12;first principles calculation;solid reaction process;electric and magnetic
