手性逆流色谱的研究进展
2016-06-22袁黎明
袁黎明
(云南师范大学化学化工学院, 云南 昆明 650500)
手性逆流色谱的研究进展
袁黎明*
(云南师范大学化学化工学院, 云南 昆明 650500)
摘要:总结了手性逆流色谱的5个特点,系统地介绍了逆流色谱的手性分离以及高速逆流色谱手性分离中氨基酸衍生物、环糊精衍生物、手性有机酸、多糖衍生物、牛血清白蛋白等手性选择剂的应用。
关键词:手性;手性拆分;逆流色谱;高速逆流色谱;综述
逆流色谱是一种不用固态支撑体或载体的液液分配色谱技术,典型特征是互不相溶的两相在分离过程中做逆流运动,溶质组分由于在两相中的分配系数不同而得到分离。一般认为逆流色谱的创始人是Ito[1], 1966年第一台逆流色谱仪问世,20世纪80年代初期高速逆流色谱出现。北京市新技术研究所张天佑研究员等[2]于20世纪70年代在我国开创了逆流色谱仪及其应用的研究。逆流色谱可分为液滴逆流色谱、旋转小室逆流色谱、离心逆流色谱3大类。离心逆流色谱又可分为两类:一类是非行星式逆流色谱,代表是匣盒式离心逆流色谱仪;另一类是行星式逆流色谱,根据分离线圈的类型、自转轴与公转轴的相对位置和角度等因素,该类仪器又可分为多种类型,代表是高速逆流色谱(HSCCC)仪。液滴逆流色谱、旋转腔室逆流色谱因为分离效率低、所需时间长,相关研究已越来越少,国内课题组也主要从事高速逆流色谱的研究。目前国内外最具代表性的相关专著主要有两本[2,3],近期最具代表性的逆流色谱综述主要有1篇[4]。图1是逆流色谱用于手性分离的论文数分布图,表1是手性选择剂的使用情况统计。
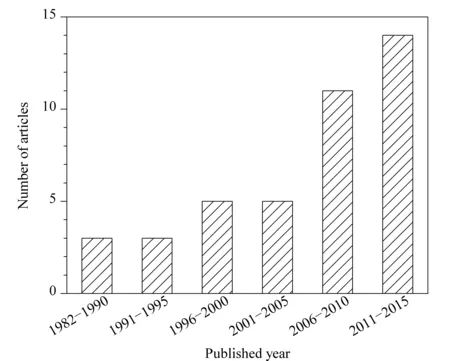
图1 逆流色谱用于手性分离的论文数分布图Fig. 1 Chart of chiral separation articles using countercurrent chromatography
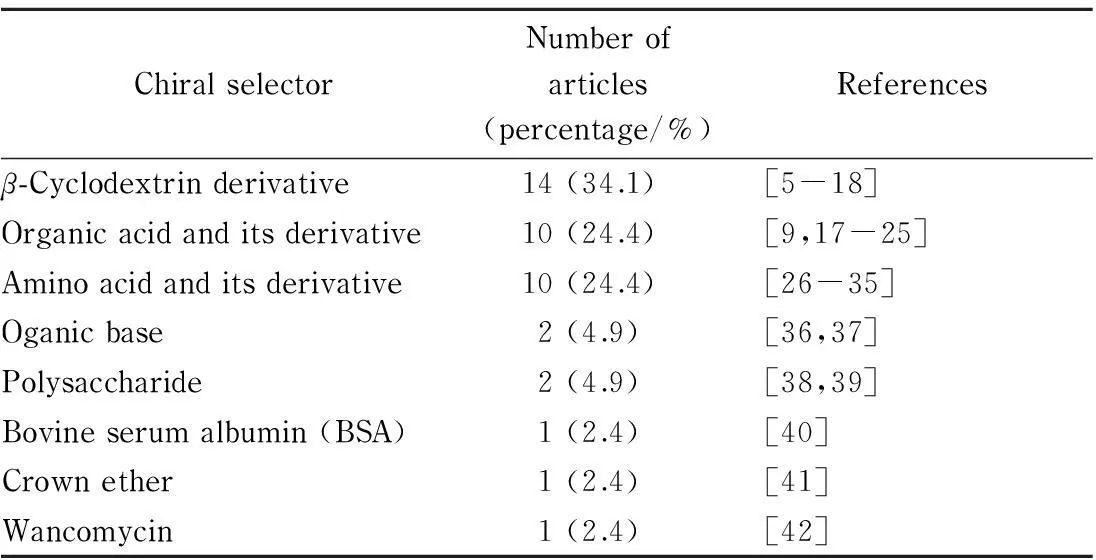
ChiralselectorNumberofarticles(percentage/%)Referencesβ-Cyclodextrinderivative14(34.1)[5-18]Organicacidanditsderivative10(24.4)[9,17-25]Aminoacidanditsderivative10(24.4)[26-35]Oganicbase2(4.9)[36,37]Polysaccharide2(4.9)[38,39]Bovineserumalbumin(BSA)1(2.4)[40]Crownether1(2.4)[41]Wancomycin1(2.4)[42]
1手性逆流色谱的特点
色谱分离法具有非常优秀的分辨能力,手性拆分属于色谱研究领域的热点和前沿,但其主要局限在高效毛细管电泳、毛细管气相色谱、高效液相色谱领域。虽然这3种方法分辨率很高,但其分离量常在微克级,只能作为分析测试手段。制备液相色谱可以满足一般性制备拆分的需要,可用于药理及毒理实验,但制备液相色谱仪价格昂贵,一根较大的手性制备柱价格可高达几万到数十万元,且大进样量严重影响柱的寿命;另外每种柱的适应范围也有限,这就造成手性制备的成本很高,即使制备较少量的手性物质,很多研究工作者都难以承受。由于逆流色谱具有制备性拆分的特点,进行逆流色谱的手性分离研究具有积极的意义。手性逆流色谱具有下面几个特点。
1.1手性选择剂在溶剂系统中的溶解度要大
手性逆流色谱要求选择的溶剂系统能分成大约相等的两相,两相有较快的分层时间(不超过30 s),能控制被拆分组分的分配比在0.5~2。另外,因为在手性逆流色谱的分离中所需加入的手性选择剂一般大于10 mmol/L,很多情况下甚至超过100 mmol/L,因此手性选择剂须在溶剂系统中有较大的溶解度。
1.2手性选择剂的选择性要高
由于逆流色谱的分离效率远低于高效液相色谱等色谱分离技术,因此在液相色谱的手性选择剂研究中,通常分离因子大于1.4的手性选择剂才有可能被成功用于逆流色谱。分离因子越大,成功的可能性越大。
1.3手性选择剂最好只溶于作为固定相的溶剂中
原则上手性选择剂可以溶解于固定相或移动相中,溶解于固定相的相当于手性柱液相色谱,溶解于移动相的相当于在流动相中含有手性添加剂的液相色谱。但要尽量选择只溶解在固定相中的手性选择剂,因为流动相中溶解的手性添加剂会大量地混合在被收集的拆分开的对映体中。
1.4手性选择剂要廉价
制备型高速逆流色谱的手性选择剂用量大,通常所配溶液浓度在10 mmol/L以上,体积在300 mL以上,所以应选择价廉的手性选择剂,否则用该方法拆分手性化合物的成本太高。
1.5产品需要进一步纯化
不管手性添加剂溶解在哪一相中,通常所收集的被拆分开的对映体中都含有手性添加剂。在逆流色谱的分离操作中,随着流动相的流出,手性选择剂会有不同程度的流失,手性选择剂流失的多少与流动相组成、流速、仪器转速、温度以及仪器类型等有关。当手性添加剂是溶解在固定相中时,手性添加剂随移动相流出的少,被拆分开的对映体中手性添加剂含量少;当手性添加剂是溶解在移动相中时,则有大量的手性添加剂混合在被拆分开的对映体中。除去被拆分开的对映体中的手性选择剂较为麻烦,在大多数情况下需要采用经典柱色谱或者重结晶的方法纯化。
2不同类型逆流色谱的手性分离应用
1982年,Hostettmann等[19]最早将逆流色谱用于手性拆分,使用旋转腔室逆流色谱仪,首次在逆流色谱中使用R,R-酒石酸-二-5-壬基酯作为手性添加剂,用1,2-二氯乙烷/水作为溶剂系统拆分了200 mg麻黄碱(±)旋光异构体。在分离过程中,0.3 mmol/L手性试剂被加在作为移动相的有机相中,水相中加入0.5 mmol/L六氟磷酸钠缓冲溶液,样品用水相溶解,移动相流速为17~20 mL/min,仪器旋转速度为60~70 r/min,实验温度为2~3 ℃和5~8 ℃,一次进样分离用时为3~4天。展示了逆流色谱对旋光异构体分离的良好潜力,只是所需时间长,分离量也不太大。
液滴逆流色谱仍有用于拆分手性化合物的示例。1984年,Takeuchi等[26]使用当时流行的液滴逆流色谱仪(含400根直径为2 cm的分离管,总容积为2 L)对亮氨酸对映异构体进行了基线分离,同时部分拆开了(±)缬氨酸和(±)蛋氨酸,用2 mmol/L合成的N-正十二烷酰基-L-脯氨酸作手性添加剂,2 mmol/L的正丁醇和1 mmol/L的醋酸铜-醋酸缓冲溶液作溶剂系统,移动相流速保持1.1 mL/min,用时2.5天。两年后,Snyder等[36]用(-)-R-2-氨基丁醇作手性添加剂,拆分了100 mg的二环[2.2.1]-庚-5-烯-2-羧酸衍生物,比传统的结晶法和酯化法拆分对映异构体的效果好。所用仪器的柱容积是280 mL,溶剂系统为pH=7的氯仿-甲醇-磷酸盐缓冲溶液(7∶13∶8, v/v/v),耗时2.3天。液滴逆流色谱仅靠流动相在重力作用下形成液滴,洗脱速率很难提高,所以耗时较长。而且单靠在分离管中简单地上升或者下降的移动相所带来的溶质在两相中的反复分配是很有限的,分离效率也不高。
Minguillón等[27]利用离心分配色谱将10 mmol/L的3,5-二甲基苯胺-N-十二烷酰基-L-脯氨酸、3,5-二甲基苯胺-N-十二烷酰基-(4R)-羟基-L-脯氨酸、3,5-二甲基苯胺-N-3,5-二甲基苯甲酰基-L-脯氨酸、3,5-二甲基苯胺-N-3,5-二甲基苯甲酰基-(4R)-羟基-L-脯氨酸分别用于3,5-二硝基苯甲酰基(DNB)-亮氨酸的手性拆分,甲基异丁基酮-0.2 mol/L磷酸盐缓冲溶液为两相溶剂系统,可以拆分开150 mg的样品。该团队[20,21]合成了(S)-萘普生的衍生物N,N-二乙基-(S)-萘普生酰胺并用作手性选择剂,采用约100 mmol/L的N,N-二乙基-(S)-萘普生酰胺,通过改变阀门方向而变化分离的正相或者反相,也实现了两个外消旋体(±)-N-(3,4-顺-3-癸基-1,2,3,4-四氢菲-4-基)-3,5-二硝基苯甲酰胺的手性分离。该团队[28]还合成了3,5-二甲基苯胺-N-全氟十二酰基-L-脯氨酸作为手性选择剂,以乙氧基九氟丁烷-异丙醇-水(25∶35∶40, v/v/v)作为两相溶剂系统,手性拆分了DNB-亮氨酸以及DNB-亮氨酸叔丁基醚。该团队[38]还将纤维素及直链淀粉的3,5-二甲基苯基氨基甲酸酯衍生物(7.5 mg/mL)用于逆流色谱中,在甲基异丁基酮-50 mmol/L磷酸盐缓冲溶液以及甲基叔丁基醚-50 mmol/L磷酸盐缓冲溶液的两相中,分别部分拆分开了40 mg品脱洛尔和50 mg华法令。该团队利用上述类似的操作条件,以甲基异丁基酮-水以及甲基叔丁基醚-水为两相溶液系统,在两相中分别加入不同的酸和碱,使用pH-区带提取技术拆分了品脱洛尔和华法令,也有一定的分离效果。为了改善纤维素在两相溶剂系统中的溶解性,该团队[39]还合成了纤维素3,5-二氯苯基氨基甲酸酯衍生物以及纤维素十二烷酰基和3,5-二甲基氨基甲酸酯的混合取代物作为手性选择剂,以离心分配色谱和pH-区带提取逆流色谱两种模式,对品脱洛尔和华法令进行了拆分,也有一定的制备性分离效果。Chung等[41]利用分析型的离心分配色谱,以少量的(+)-(18-冠-6)-四羧酸为手性选择剂,实现了对抗菌药物吉米沙星的手性分离。
金刚烷基氨基甲酰基奎宁和金刚烷基氨基甲酰基奎宁啶是两种有效的离子交换色谱的手性添加剂,它们易于接受和失去质子,在分离氨基酸时立体选择性很好。Lindner等[37]通过使用离心式逆流色谱,用pH 6.0的0.1 mmol/L氨基乙酸缓冲液-叔戊基醇-甲醇-庚烷(10∶5∶1∶5, v/v/v/v)作溶剂系统,含10.6 mmol/L的手性添加剂,转速为1 000 r/min,流速为3 mL/min,很好地分离了DNB-亮氨酸、3,5-二硝基苯甲氧酰基(DNZ)-叔戊基甘氨酸和DNZ-β-苯丙氨酸。用pH 8.0的0.1 mol/L氨基乙酸缓冲液-丙酮-甲基异丁基酮(MIBK)(2∶1∶2, v/v/v)最多分离了300 mg的DNB-亮氨酸。用pH-区带提取逆流色谱,以含三氟乙酸(10 mmol/L)的金刚烷基氨基甲酰基奎宁的MIBK溶液作固定相,用220 mmol/L氨水作移动相,转速为1 200 r/min,流速为3 mL/min,最多分离了900 mg的DNB-亮氨酸。
Duret等[42]在用行星式逆流色谱仪进行手性拆分实验时,选用甲苯和水作溶剂系统,使用在液相色谱、薄层色谱和毛细管电泳中有效的手性添加剂万古霉素(pH 4.7, 140 mg/mL),拆分了D,L-丹磺酰正亮氨酸。分离过程中,当从尾到头泵入移动相时,左消旋体先被洗脱出来;当反向泵入移动相时,右消旋体先被洗脱出来。结果表明万古霉素也适用于作逆流色谱的手性添加剂,而且最多能分离50 mg的D,L-丹磺酰正亮氨酸。
3高速逆流色谱的手性分离应用
逆流色谱拆分手性化合物的研究已经走过了30余年,本课题组在国家自然科学基金的资助下,于1997年开展了高速逆流色谱制备性分离植物生物碱及黄酮的研究(No.29665001),于2002年率先在国内开展了高速逆流色谱的手性分离研究(No.30160092)。目前国内已有多个团队从事该领域的研究,近5年来在国际学术期刊上发表的逆流色谱用于手性分离的论文主要来自于我国。使用逆流色谱拆分手性化合物的代表性综述大约有3篇[43-45]。
3.1氨基酸及其衍生物
氨基酸衍生物类手性固定相广泛应用于高效液相色谱研究中,Foucault等[29]使用HSCCC,将添加剂3,5-二甲基苯胺-N-十二烷酰基-L-脯氨酸加入庚烷-乙酸乙酯-甲醇-水(3∶1∶3∶1, v/v/v/v)溶剂系统中,较快(80 min)拆分开了两种氨基酸衍生物DNB-叔丁基缬氨酸酰胺和DNB-叔丁基亮氨酸酰胺。随后,Ito等[30,31]使用这类具有π电子的手性添加剂,同样使用HSCCC,选用了两种溶剂系统:正己烷-乙酸乙酯-甲醇-10 mmol/L盐酸(8∶2∶5∶5, v/v/v/v)和正己烷-乙酸乙酯-甲醇-10 mmol/L盐酸(6∶4∶5∶5, v/v/v/v),成功分离了(±)DNB-苯甘氨酸和(±)DNB-苯丙氨酸等。通过改变手性添加剂在固定相中的浓度,一次进样最多能分离1 g的手性样品。
颜继忠等[32]利用分析型高速逆流色谱,丁醇-水(1∶1, v/v)或者正己烷-丁醇-水(0.5∶0.5∶1, v/v/v)为两相溶剂系统,将50 mmol/LN-十二烷酰基-L-脯氨酸作为手性选择剂添加到有机相中,水相中加入25 mmol/L醋酸铜,系统地研究了其对扁桃酸、2-氯扁桃酸、4-甲氧基扁桃酸、4-羟基扁桃酸、α-甲基扁桃酸、4-羟基-3-甲氧基扁桃酸、3-氯扁桃酸、4-溴扁桃酸、α-环戊基扁桃酸和α-环己基扁桃酸的手性拆分,结果扁桃酸、2-氯扁桃酸、4-甲氧基扁桃酸、4-羟基扁桃酸、α-甲基扁桃酸、4-羟基-3-甲氧基扁桃酸、3-氯扁桃酸得到不同程度的分离。将仪器改为制备型的高速逆流色谱仪后,其最大拆分量可以达到数十毫克。由于醋酸铜的加入,手性配位参与了手性识别过程,因此该手性分离属于手性配体交换色谱分子作用机理。
魏云等[33]将脯氨酸衍生成为手性离子液体1-乙基-3-甲基-咪唑-L-脯氨酸并将其键合在磁纳米球的表面,以乙醇-水或者正丁醇-水(1∶1, v/v)为两相溶剂系统,用600 mg的该手性材料拆分了0.75 mg的色氨酸的外消旋体。
氨基酸衍生物还可用于pH-区带提取逆流色谱技术的手性拆分[46]。在少量酸性物质的样品溶液中加入一定浓度有机酸时,可产生一个特殊的窄而尖的峰。当样品量增大时,每个组分将在柱中形成一个高度浓缩的等pH区带,并以一个矩形峰被洗脱出来;当样品量继续增大,矩形峰也随着增宽,但并不影响组分之间的分离效果。这就使该方法能在一般高速逆流色谱法的基础上成十倍地增加样品进样量,而不用对常见设备做任何改进。Ito等[34,35]在固定相中添加N-十二烷酰基-L-脯氨酸和三氟乙酸,在移动相添加中氨水,用330 mL容积的高速逆流色谱仪,经过3 h分离了2 g的(±)DNB-亮氨酸。这是目前一次性分离手性化合物的量最大的研究论文,pH-区带提取逆流色谱也因此成为分离量最大的制备性分离手性化合物的逆流色谱技术。
3.2环糊精及其衍生物
环糊精及其衍生物是应用范围广泛的手性选择剂。最先成功用于逆流色谱的是磺化-β-环糊精,研究人员[5]在高速逆流色谱上做了大量探索实验,最终选用乙酸乙酯-甲醇-水(10∶1∶9, v/v/v)作溶剂系统,水相中含2%的磺化-β-环糊精作为手性添加剂,基线分离了(±)-7-去甲基-奥美昔芬。2010年,Wei等[6]将50 mmol/L磺化-β-环糊精作为手性选择剂,乙酸乙酯-甲醇-水(10∶1∶10, v/v/v)为两相溶剂系统,一次性拆分了20 mg的抗菌药物洛美沙星,为逆流色谱手性添加剂的选择打开了思路。
本课题组[7]较早应用羧甲基环糊精成功地对扑尔敏进行了制备性分离,溶剂系统为乙酸乙酯-甲醇-水(10∶1∶9, v/v/v)。当羧甲基-β-环糊精在固定相中的浓度为20 mmol/L时,获得了较好的对映异构体的分离效果。手性添加剂过少或过多都不利于外消旋体的拆分。这是国内学者在国外发表的第一篇利用逆流色谱制备性拆分手性化合物的学术论文,本课题组当时也在国内对手性逆流色谱的进展进行了介绍[44,47]。
本课题组[8]还以羧甲基-β-环糊精对氨鲁米特外消旋体进行了制备性分离,溶剂系统为乙酸乙酯-甲醇-水(10∶1∶9, v/v/v)。羧甲基-β-环糊精在固定相中的浓度为20 mmol/L,一次拆分了3 mg氨鲁米特外消旋体药物。
颜继忠课题组[9-13]将羟丙基-β-环糊精用于高速逆流色谱研究,拆分了具有类似苯丙酸母体结构的手性化合物。它们分别是α-环己基苯乙酸、萘普生、苯基丁二酸和苯丙酸,分子结构如图2所示。在这些拆分中,溶剂系统分别采用了正己烷-甲基叔丁基醚-水(9∶1∶10, v/v/v)、正己烷-乙酸乙酯-0.1 mol/L磷酸缓冲溶液(8∶2∶10、7.5∶2.5∶10, v/v/v)、正己烷-甲基叔丁基醚-0.1 mol/L磷酸缓冲溶液(0.5∶1.5∶2, v/v/v)和正己烷-乙酸乙酯-0.1 mol/L磷酸缓冲溶液(8∶2∶10、5∶5∶10、7∶3∶10, v/v/v),所用羟丙基-β-环糊精的浓度为100~300 mmol/L,一次拆分外消旋体的量多数在几十毫克数量级,量大的达到712 mg。从他们的研究可以看出,羟丙基-β-环糊精对于芳丙酸类似物的手性拆分是非常有效的。
奥昔布宁(图2)是一种治疗尿路疾病的药物,也具有苯丙酸的分子骨架结构。Tang等[14]以正己烷-甲基叔丁基醚-0.1 mol/L磷酸盐缓冲溶液(6∶4∶10, v/v/v)为溶剂系统,100 mmol/L羟丙基-β-环糊精为手性选择剂,一次可拆分15 mg的样品。
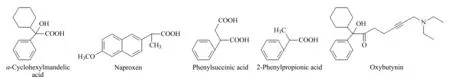
图2 芳丙酸类似物分子结构图Fig. 2 Molecular structures of phenylpropionic acid derivatives
孔令仪团队[15]也将25 mmol/L羟丙基-β-环糊精用于反式-δ-葡萄素的手性拆分,溶剂系统是正己烷-乙酸乙酯-水(5∶5∶10, v/v/v),一次进样拆分了20 mg的反式-δ-葡萄素对映异构体。最近,该团队将双核Cu2(Ⅱ)-β-环糊精用于α-环己基扁桃酸的拆分,溶剂系统为正己烷-乙酸乙酯-0.1 mol/L磷酸缓冲溶液(9∶1∶10, v/v/v), Cu2(Ⅱ)-β-环糊精的浓度为40 mmol/L,一次进样拆分了10 mg样品。双核Cu2(Ⅱ)-β-环糊精的拆分效果明显优于未衍生化的β-环糊精。在其他色谱技术中将双核Cu2(Ⅱ)-β-环糊精用于手性拆分的报道较少,更可贵的是在相同的实验条件下,还同时拆分了芳丙酸类似物扁桃酸、α-环戊基扁桃酸、α-甲基扁桃酸、4-甲氧基扁桃酸和4-羟基扁桃酸[16]。
3.3手性有机酸
本课题组[22]应用L-(+)-酒石酸制备性拆分了氧氟沙星,溶剂系统为乙酸乙酯-甲醇-水(10∶1∶9, v/v/v),手性选择剂L-酒石酸在固定相中的浓度为200 mmol/L。还应用L-(+)-酒石酸一次性拆分了120 mg的α-甲基苯胺,溶剂系统为氯仿-甲醇-水(4∶3∶1, v/v/v), L-(+)-酒石酸在固定相中的浓度为278 mmol/L[23]。
颜继忠等[24]将二正己基酒石酸(100 mmol/L)作为手性选择剂,以氯仿-0.05 mol/L醋酸盐缓冲溶液(含硼酸100 mmol/L)(1∶1, v/v)作为溶剂系统,能制备性拆分β-受体阻滞剂普萘洛尔、品脱洛尔以及阿普洛尔。该研究不但能拆分多个β-受体阻滞剂药物,而且还利用pH-区带提取逆流色谱技术,通过在有机相中加入三乙胺,在水相中加入盐酸,一次性拆分了356 mg的普萘洛尔外消旋体。该团队还将二正丁基酒石酸(100 mmol/L)作为手性选择剂,以氯仿-0.05 mol/L醋酸盐缓冲溶液(含硼酸100 mmol/L)(1∶1, v/v)为溶剂系统,拆分了92 mg的另一个β-受体阻滞剂药物丙胺苯丙酮[25]。
Tang等[17]对酒石酸衍生物作为手性萃取剂进行了较多的研究,近年来成功地将其与环糊精衍生物混合应用于高速逆流色谱的手性分离研究。将异丁基酒石酸(50 mol/L)与羟丙基-β-环糊精(50 mol/L)作为手性选择剂,正己烷-甲基叔丁基醚-0.1 mol/L磷酸盐缓冲液(0.5∶1.5∶2, v/v/v)为溶剂系统,进样体积20 mL,一次性分离了810 mg的芳丙酸类似物苯基丁二酸。该团队还将异丁基酒石酸(100 mol/L)与磺丁基-β-环糊精(50 mol/L)作为手性选择剂,乙酸乙酯-水(1∶1, v/v)为两相溶剂系统,一次性进样后通过循环高速逆流色谱分离了30 mg的心血管药物苯磺酸氨氯地平[18]。
另外,本课题组尚未发表的研究结果也显示利用樟脑磺酸作手性添加剂对拉贝洛尔外消旋体有一定的制备性拆分效果。
3.4牛血清白蛋白
Ito等[40]先使用旋转腔室逆流色谱仪,用牛血清白蛋白(BSA)作手性添加剂拆分D,L-犬尿氨酸,用时60 h仍没有得到基线分离;后改用高速逆流色谱,10%(质量分数,下同)的PEG 800作固定相,5%磷酸二氢钠缓冲溶液和6%BSA作移动相,转速800 r/min,流速0.2 mL/min,只耗时3.5 h就成功拆分2.5 mg D,L-色氨酸。说明高速逆流色谱的分离能力比旋转腔室逆流色谱更强,也证实了牛血清白蛋白能用于逆流色谱的手性拆分。
4小结
逆流色谱具有制备性拆分的优点,1982年就开始了其对手性化合物的拆分研究,但总的来讲逆流色谱的手性拆分研究发展缓慢。已报道的用于逆流色谱的手性分离材料种类仍然非常有限,早期被拆分的物质主要是氨基酸的衍生物,整个逆流色谱领域就作者掌握的手性拆分文献仅有40多篇。该技术的应用在2010年后有明显的增加趋势,但被拆分的对映体较集中在芳基丙酸母体结构的化合物以及氨基酸的衍生物。由于该方法手性材料选择困难,每一次拆分手性材料的用量较大,尤其是被拆分开的对映体中往往含有一定量甚至是大量的手性试剂而不得不通过柱色谱或者结晶法进一步提纯,以及现代手性制备液相色谱的突飞猛进等多种原因,使该手性分离方法的实际应用还很少,所有这些都还需要去解决,使其能真正应用于科研及生产实践中。
参考文献:
[1]Ito Y, Weinstein M A, Aoki I, et al. Nature, 1966, 212: 985
[2]Zhang T Y, Wang X. High Speed Countercurrent Chromatography. Beijing: Chemical Industry Press, 2011
张天佑, 王晓. 高速逆流色谱技术. 北京: 化学工业出版社, 2011
[3]Ito Y, Conway W D. High-Speed Countercurrent Chromatography. New York: John Wiley & Sons Inc., 1996
[4]Pan Y J, Lu Y B. J Liq Chromatogr R T, 2007, 30: 649
[5]Breinholt J, Lehmann S V, Varming A R. Chirallity, 1999, 11: 768
[6]Wei Y, Du S, Ito Y. J Chromatogr B, 2010, 878: 2937
[7]Yuan L M, Liu J C, Yan Z H, et al. J Liq Chromatogr, 2005, 28: 3057
[8]Ai P, Liu J C, Zi M, et al. Chinese Chemical Letters, 2006, 17(6): 787
[9]Tong S, Yan J, Guan Y X, et al. J Chromatogr A, 2010, 1217: 3044
[10]Tong S, Guan Y X, Yan J, et al. J Chromatogr A, 2011, 1218: 5434
[11]Tong S, Yan J, Guan Y X, et al. J Chromatogr A, 2011, 1218: 5602
[12]Tong S, Zheng Y, Yan J. J Chromatogr A, 2013, 1281: 79
[13]Tong S, Zheng Y, Yan J. J Sep Sci, 2013, 36: 2035
[14]Zhang P, Sun G, Tang K, et al. J Sep Sci, 2014, 37: 3443
[15]Han C, Xu J, Wang X, et al. J Chromatogr A, 2014, 1324: 164
[16]Han C, Luo J, Xu J, et al. J Chromatogr A, 2015, 1375: 82
[17]Sun G, Tang K, Zhang P, et al. J Sep Sci, 2014, 37: 1736
[18]Zhang P, Sun G, Tang K, et al. Sep Purif Technol, 2015, 146: 276
[19]Domon B, Hostettmann K, Kovacevic K, et al. J Chromatogr, 1982, 250: 149
[20]Rubio N, Ignatova S, Minguillón C, et al. J Chromatogr A, 2009, 1216: 8505
[21]Rubioa N, Minguillón C. J Chromatogr A, 2010, 1217: 1183
[22]Lv Y C, Yan Z H, Ma C, et al. J Liq Chromatogr R T, 2010, 33: 1328
[23]Cai Y, Yan Z H, Zi M, et al. J Liq Chromatogr R T, 2007, 30: 1489
[24]Tong S, Zheng Y, Yan J, et al. J Chromatogr A, 2012, 1263: 74
[25]Tong S, Zheng Y, Yan J. J Sep Sci, 2013, 36: 2035
[26]Takeuchi T, Horikawa R, Tanimura T. J Chromatogr, 1984, 284: 285
[27]Delgado B, Pérez E, Santano M C, et al. J Chromatogr A, 2005, 1092: 36
[28]Pérez A M, Minguillón C. J Chromatogr A, 2010, 1217: 1094
[29]Oliveros L, Puertolas P F, Minguillon C, et al. J Liq Chromatogr, 1994, 11: 2301
[30]Ma Y, Ito Y. Anal Chem, 1995, 67(17): 3069
[31]Ma Y, Ito Y, Berthod A. J Liq Chromatogr, 1999, 22: 2945
[32]Tong S, Shen M, Cheng D, et al. J Chromatogr A, 2014, 1360: 110
[33]Liu Y, Tian A, Wang X, et al. J Chromatogr A, 2015, 1400: 40
[34]Ma Y, Ito Y, Foucault A. J Chromatogr A, 1995, 704: 75
[35]Ma Y, Ito Y. Anal Chem, 1996, 68(7): 1207
[36]Oya S, Snyder J K. J Chromatogr, 1986, 370: 333
[37]Franco P, Blanc J, Oberleitner W R, et al. Anal Chem, 2002, 74(16): 4175
[38]Pérez E, Santos M J, Minguillón C. J Chromatogr A, 2006, 1107: 165
[39]Pérez E, Minguillón C. J Sep Sci, 2006, 29: 1379
[40]Shinomiya K, Kabasawa K, Ito Y. J Liq Chromatogr, 1998, 21: 135
[41]Kim E, Koo Y M, Chung D S. J Chromatogr A, 2004, 1045: 119
[42]Duret P, Foucault A, Margraff R. J Liq Chromatogr, 2000, 23: 295
[43]Foucault A P. J Chromatogr A, 2001, 906: 365
[44]Yan Z H, Ai P, Yuan L M, et al. Chinese Journal of Chemistry, 2006, 69(1): 33
严志宏, 艾萍, 袁黎明, 等. 化学通报, 2006, 69(1): 33
[45]Huang X Y, Di D L. TrAC-Trends Anal Chem, 2015, 67: 128
[46]Weisz A, Scher A L, Shinomiya K, et al. J Am Chem Soc, 1994, 116(2): 704
[47]Yuan L M. Preparative Chromatography Technology and Application. Beijing: Chemical Industry Press, 2005: 132
袁黎明. 制备色谱技术及应用. 北京: 化学工业出版社, 2005: 132
Progress of chiral countercurrent chromatography
YUAN Liming*
(School of Chemistry and Chemical Engineering, Yunnan Normal University, Kunming 650500, China)
Abstract:This article summarizes five characteristics of chiral countercurrent chromatography (CCC). The progress of chiral countercurrent chromatography is introduced. In the high speed countercurrent chromatography, the chiral selectors such as amino acid derivatives, cyclodextrin derivatives, chiral organic acid, polysaccharide and bovine serum albumin (BSA) are reviewed.
Key words:chirality; enantioseparation; countercurrent chromatography (CCC); high speed countercurrent chromatography (HSCCC); review
DOI:10.3724/SP.J.1123.2015.09013
*收稿日期:2015-09-13
基金项目:国家自然科学基金项目(21275126).
中图分类号:O658
文献标识码:A
文章编号:1000-8713(2016)01-0044-06
色谱手性分离专刊·专论与综述
*通讯联系人.Tel:(0871)65941088,E-mail:yuan_limingpd@126.com.
Foundation item: National Nature Science Foundation of China (Grant No. 21275126).
