新型2-(吡咯烷-3-基)-噁唑衍生物的合成
2016-06-13李翠平马珍珍刘艳玲孙春玲
李翠平, 马珍珍, 刘艳玲, 樊 振, 孙春玲, 王 伟
(天方药业有限公司,河南 驻马店 463000)
·研究简报·
新型2-(吡咯烷-3-基)-噁唑衍生物的合成
李翠平, 马珍珍, 刘艳玲, 樊振, 孙春玲, 王伟*
(天方药业有限公司,河南 驻马店463000)
摘要:根据亚结构链接法,以氯甲基三甲基硅烷和苄胺为原料,依次经酯化、缩合、环合、水解、成盐等12步反应制得5个新型的(4-甲基氨基甲酰基-2-吡咯烷-5-甲基噁唑)-胺基甲酸叔丁酯衍生物(15a~15e); 15a与醛(酮)或乙酰氯反应,合成了4个新型的5-胺基甲基-2-(1-甲基-吡咯烷)-噁唑-N-甲基甲酰胺盐酸盐衍生物,其结构经1H NMR和ESI-MS表征。
关键词:亚结构链接法; 甲基氨基甲酰基; 噁唑; 吡咯烷; 合成
噁唑环是一类重要的五元氮氧杂环,具有易形成氢键、与金属离子配位以及产生疏水作用、π-π堆积、静电作用等特点。因此,噁唑类化合物可产生多种非共价键相互作用[1],显示出良好的杀虫、除草和抗菌等生物活性[2-5]。噁唑类化合物广泛应用于制药领域,如除草剂双苯噁唑酸[6]、临床抗菌药物利奈唑胺[7]和维吉霉素[8]等。
吡咯烷是一类五元氮杂环生物碱化合物[9],其环张力较小,稳定性较好,是多种药物的成份和重要的化工中间体[10]。吡咯烷衍生物的合成及其生物活性研究越来越受到人们的重视。
亚结构链接法可将具有不同生物活性的杂环拼接至同一分子中,得到高活性的新型化合物。根据该原理,本文以氯甲基三甲基硅烷(1)和苄胺为原料,依次经酯化、缩合、环合、水解、成盐等12步反应制得5个新型的(4-甲基氨基甲酰基-2-吡咯烷-5-甲基噁唑)-胺基甲酸叔丁酯衍生物(15a~15e); 15a与醛(酮)或乙酰氯反应,合成了4个新型的5-胺基甲基-2-(1-甲基-吡咯烷)-噁唑-N-甲基甲酰胺盐酸盐衍生物(18f~18i, Scheme 1),其结构经1H NMR和ESI-MS表征。
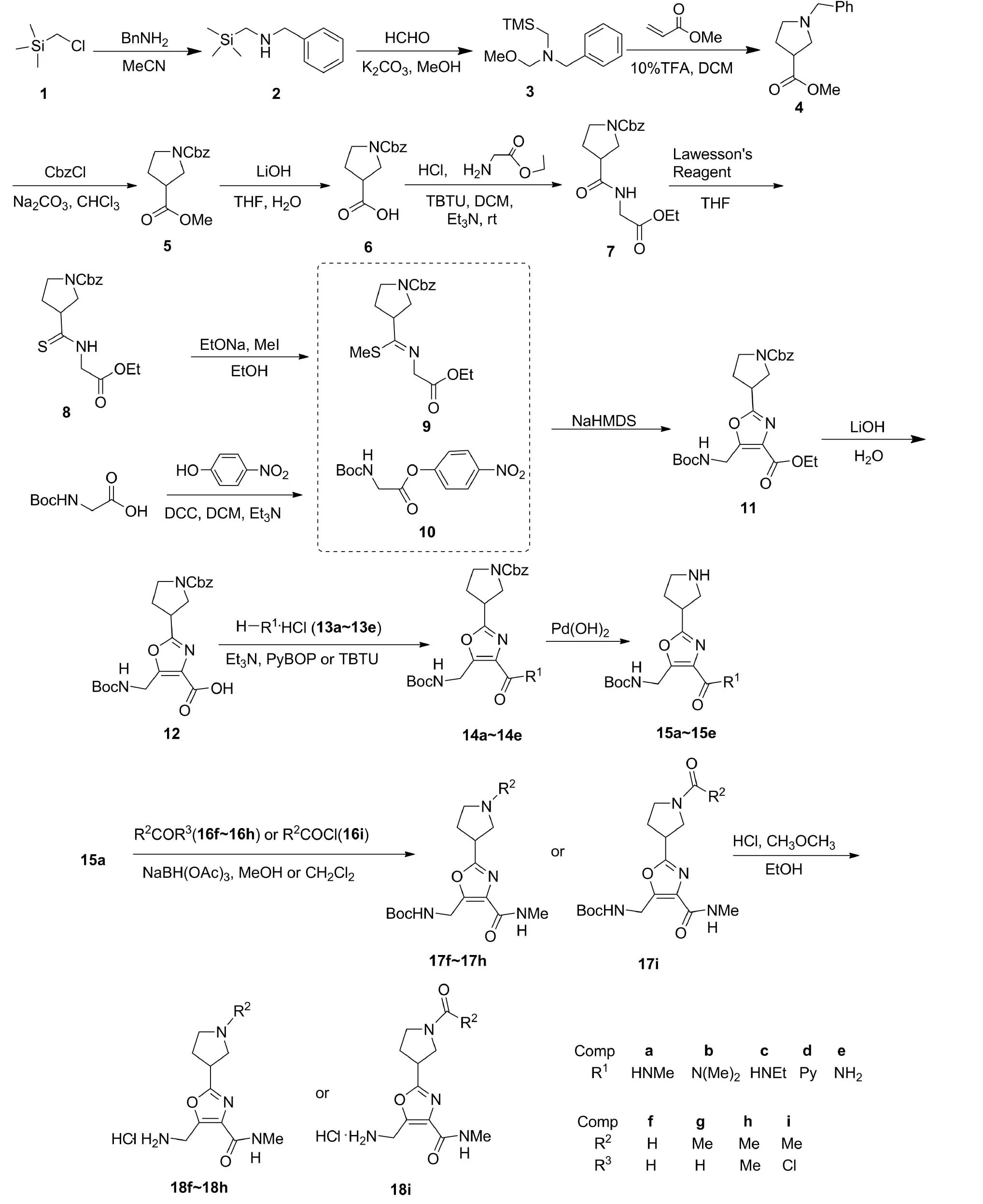
Scheme 1
1实验部分
1.1试剂与仪器
Bruker AV 400型核磁共振仪(CDCl3为溶剂,TMS为内标);Waters Acquity型液质联用仪。
所用试剂均为分析纯。
1.2合成
(1) 苄基三甲基硅烷基甲基胺(2)的合成
在反应瓶中加入1 25.0 g(0.2 mol),苄胺44.0 g(0.4 mol)和乙腈200 mL,回流反应20 h(TLC检测)。浓缩,残余物用水洗涤,用乙酸乙酯(3×100 mL)萃取,合并有机相,浓缩得淡黄色液体2 32.7g,收率83%;1H NMRδ: 7.41~7.31(m, 5H), 3.87(s, 2H), 2.13(s, 2H), 0.12(s, 9H)。
(2) 苄基甲氧基甲基三甲基硅烷基甲基胺(3)的合成
在反应瓶中加入2 32.7 g(169 mmol), 50%甲醛溶液12.2 g(203 mmol)和甲醇100 mL,搅拌下于0 ℃缓慢加入碳酸钾20.4 g,加毕(0.5 h),于室温反应5 h。抽滤,滤液浓缩得淡黄色液体3 38.2 g,收率95%;1H NMRδ: 7.37~7.27(m, 5H), 4.05(s, 2H), 3.39(s, 2H), 3.28(s, 3H), 2.23(s, 2H), 0.09(s, 9H)。
(3) 1-苄基吡咯烷-3-羧酸甲酯(4)的合成
在反应瓶中加入3 38.2 g(160 mmol),丙烯酸甲酯20.8 g(240 mmol)和二氯甲烷100 mL,搅拌下于0 ℃缓慢滴加10%三氟乙酸12.0 g(10 mmol),滴毕(1 h),于室温反应2 h。减压浓缩,剩余物经硅胶柱层析[洗脱剂A:V(石油醚) ∶V(乙酸乙酯)=10 ∶1]纯化得白色固体4 30.3 g,收率86%;1H NMRδ: 7.35~7.27(m, 5H), 5.12(s, 2H), 3.69~3.56(m, 6H), 3.41~3.44(m, 1H), 3.02~3.09(m, 1H), 2.14(t,J=6.7 Hz, 2H)。
(4) 吡咯烷-1,3-二甲酸-1-苄酯-3-甲酯(5)的合成
在反应瓶中加入4 30.3 g(137 mmol),氯甲酸苄酯47.2 g(275 mmol),无水碳酸钠43.0 g和氯仿300 mL,搅拌下回流(62 ℃)反应5 h。减压浓缩得淡黄色固体5 32.8 g,收率90%;1H NMRδ: 7.38~7.32(m, 5H), 5.15(s, 2H), 3.72~3.50(m, 6H), 3.49~3.41(m, 1H), 3.06(m, 1H), 2.19~2.14(m, 2H)。
(5) 吡咯烷-1,3-二甲酸-1-苄酯(6)的合成
0 ℃下向反应瓶中加入5 32.8 g(123 mmol),氢氧化锂30.4 g(1.233 mol), THF和水的混合溶液[V(THF) ∶V(水)=1 ∶1]500 mL,反应1 h(TLC检测)。于室温用0.5 mol·L-1盐酸调至pH 5~6,用二氯甲烷(3×150 mL)萃取,合并有机相,减压浓缩得淡黄色固体6 28.8 g,收率98%;1H NMRδ: 7.40~7.32(m, 5H), 5.17(s, 2H), 3.73~3.68(m, 2H), 3.60~3.57(m, 1H), 3.50~3.48(m, 1H), 3.14~3.11(m, 1H), 2.22~2.17(m, 2H)。
(6) 3-(乙氧羰基甲基-氨基甲酰基)-吡咯烷-1-甲酸苄酯(7)的合成
在反应瓶中加入6 30 g(120 mmol),氨基乙酸乙酯盐酸盐16.4 g(132 mmol),三乙胺36.7 g(367 mmol),O-苯并三氮唑-N,N,N′,N′-四甲基脲四氟硼酸(TBTU)42.5 g和二氯甲烷250 mL,于室温反应2 h。用0.5 mol·L-1盐酸调至pH 7,分液,有机相减压浓缩后经硅胶柱层析(洗脱剂:A=10∶1)纯化得黄色固体7 33.5 g,收率84%;1H NMRδ: 7.50~7.30(m, 5H), 5.14(s, 2H), 4.23(q,J=7.2 Hz, 2H), 4.04(s, 2H), 3.80~3.50(m, 2H), 3.50~3.40(m, 1H), 3.05~2.90(m, 2H), 2.16~2.05(m, 2H), 1.29(t,J=7.2 Hz, 3H)。
(7) 3-(乙氧羰基甲基-硫代氨基甲酰基)-吡咯烷-1-甲酸苄酯(8)的合成
在反应瓶中加入7 30 g(90 mmol),劳森试剂25 g(60 mmol)和THF 300 mL,搅拌下回流反应3 h。减压浓缩,剩余物经硅胶柱层析(洗脱剂:A=8 ∶1)纯化得黄色固体8 24 g,收率76%;1H NMRδ: 7.40~7.30(m, 5H), 5.30(s, 2H), 4.38(s, 2H), 4.27(q,J=7.2 Hz, 2H), 3.85~3.75(m, 1H), 3.75~3.65(m, 2H), 3.47~3.40(m, 1H), 3.35~3.20(m, 1H), 2.40~2.30(m, 1H), 2.20~2.10(m, 1H), 1.31(t,J=7.2 Hz, 3H)。
(8) 3-(乙氧羰基甲基亚胺基-甲基磺酰基-甲基)-吡咯烷-1-甲酸苄酯(9)的合成
在反应瓶中加入8 30 g(85 mmol)和乙醇300 mL,搅拌下于-20 ℃缓慢滴加20%乙醇钠(30 g)的乙醇溶液,滴毕(1 h),加入碘甲烷37 g(262 mmol),于室温反应过夜。用磷酸氢钾饱和溶液(200 mL)淬灭反应,蒸除乙醇,残余物用蒸馏水(150 mL)洗涤,用乙酸乙酯(3×120 mL)萃取,合并有机相,用无水硫酸钠干燥,浓缩,剩余物经硅胶柱层析(洗脱剂:A=8∶1)纯化得淡黄色固体9 27.2 g,收率90%;1H NMRδ: 7.45~7.37(m, 5H), 5.15(s, 2H), 4.23(s, 2H), 3.92(q,J=7.2 Hz, 2H), 3.65~3.61(m, 1H), 3.59~3.51(m, 2H), 3.47~3.40(m, 1H), 3.31~3.25(m, 1H), 2.76~2.71(m, 3H), 2.36~2.34(m, 1H), 2.25~2.22(m, 1H), 1.31(t,J=7.2 Hz, 3H)。
(9) Boc-甘氨酸-对硝基苯酯(10)的合成
在反应瓶中依次加入二氯甲烷1 L, Boc-甘氨酸100.0 g(570 mmol),对硝基苯酚79.0 g(570 mmol)和二环己基碳二亚胺117.4 g(570 mmol),搅拌下缓慢滴加三乙胺115.0 g(1.14 mol),滴毕,反应18 h(TLC检测)。反应液用饱和碳酸钠溶液洗涤,干燥,浓缩,残余物经硅胶柱层析(洗脱剂:A=2 ∶1)纯化得白色固体10 100 g,收率91%;1H NMRδ: 8.30(d,J=8.8 Hz, 2H), 7.34(d,J=9.2 Hz, 2H), 4.22(d,J=5.2 Hz, 2H), 1.49(s, 9H)。
(10) 2-(1-苄氧羰基-吡咯烷)-5-(叔丁氧羰基氨基-甲基)-噁唑-4-羧酸乙酯(11)的合成
在反应瓶中加入9 30 g(82 mmol)和THF 300 mL,搅拌下于-78 ℃依次缓慢滴加NaHMDS 16 g的THF(200 mL)溶液(10 min), 10 30 g(98 mmol)的THF(100 mL)溶液和NaHMDS 16 g(95 mmol)的THF(200 mL)溶液,滴毕,反应5 h。用0.5 mol·L-1盐酸淬灭反应,于室温用乙酸乙酯(3×120 mL)萃取,合并有机相,减压浓缩,残余物经硅胶柱层析(洗脱剂:A=3 ∶1)纯化得白色固体11 22.0 g,收率55%;1H NMRδ: 7.37~7.28(m, 5H), 5.15(s, 2H), 4.64(s, 2H), 4.43~4.40(q,J=7.2 Hz, 2H), 3.90~3.81(m, 1H), 3.75~3.45(m, 4H), 2.37~2.30(m, 2H), 1.45(s, 9H), 1.41(t,J=7.2 Hz, 3H); ESI-MSm/z: {[M+H]+} 474.2。
(11) 2-(1-苄氧羰基-吡咯烷)-5-(叔丁氧羰基胺基-甲基)-噁唑-4-羧酸(12)的合成
在反应瓶中加入11 20 g(420 mmol)和甲醇200 mL,搅拌使其溶解;于0 ℃缓慢滴加LiOH·H2O 7 g的水(200 mL)溶液,滴毕,于室温反应至终点(TLC检测)。用0.5 mol·L-1盐酸调至pH 5~6,用乙酸乙酯(3×20 mL)萃取,合并有机相,蒸除溶剂得白色固体12 13.5 g,收率73%;1H NMRδ: 7.36~7.30(m, 5H), 5.12(s, 2H), 4.57(m, 2H), 3.87(m, 1H), 3.72~3.52(m, 4H), 2.34(m, 2H), 1.43(s, 9H); ESI-MSm/z: {[M+H]+}446.1。
(12) 15a~15c的合成(以15a为例)
在反应瓶中依次加入DMF 1 L, 12 50.0 g(0.11 mol), TBTU 54 g,甲胺盐酸盐(13a)9.4 g(0.14 mol)和Et3N 34 g,于室温反应过夜(TLC检测)。反应液用水洗涤,再用DCM萃取,有机相浓缩后经硅胶柱层析(洗脱剂:A=5 ∶1)纯化得黄色油状液体3-[5-(叔丁氧羰基胺基-甲基)-4-甲基甲酰胺基-噁唑]-吡咯烷-1-甲酸苄酯(14a),直接投入下步反应。
在反应釜中加入14a, 20%Pd(OH)2/C 14.0 g和甲醇350 mL,通入氢气(0.3 MPa),于室温反应2 h。抽滤,滤液浓缩得白色固体(4-甲基胺基甲酰基-2-吡咯烷-5-甲基-噁唑)-胺基甲酸叔丁酯(15a)。
以13b和13c替代13a,用类似的方法合成白色固体[(4-(二甲基胺基甲基)-2-(吡咯烷-3-基)噁唑-5-基)甲基]胺基甲酸叔丁酯(15b)和{[4-(乙基胺基甲酰基)-2-(吡咯烷-3-基)噁唑-5-基]甲基}胺基甲酸叔丁酯(15c)。
15a: 收率68%;1H NMRδ: 4.44(s, 2H), 3.31~3.24(m, 1H), 3.12~3.07(m, 1H), 2.89~2.80(m, 3H), 2.70(s, 3H), 2.05~1.98(m, 1H), 1.96~1.88(m, 1H), 1.44(s, 9H); ESI-MSm/z: {[M+H]+}325.2。
15b: 收率74%;1H NMRδ: 4.50(s, 1H), 5.94(s, 2H), 3.46~3.42(m, 1H), 3.35(s, 3H), 3.32~3.16(m, 3H), 3.08~3.04(m, 4H), 2.30~2.10(m, 2H), 1.46(s, 9H); ESI-MSm/z: {[M+H]+}339.1。
15c: 收率77%;1H NMRδ: 7.23(s, 1H), 6.12(s, 1H), 5.31(s, 1H), 4.58(d,J=6.0 Hz, 2H), 3.51~3.47(m, 1H), 3.47~3.42(m, 2H), 3.40~3.30(m, 2H), 3.30~3.20(m, 1H), 3.20~3.11(m, 1H), 2.24~2.30(m, 1H), 2.20~2.10(m, 1H), 1.44(s, 9H), 1.25(t,J=7.2 Hz, 3H); ESI-MSm/z: {[M+H]+}339.2。
(13) 15d和15e的合成(15d为例)
搅拌下,在反应瓶中依次加入无水DMF 250 mL, 12 23.0 g(51.6 mmol),六氟磷酸苯并三唑-1-基-氧基三吡咯烷基(PyBOP)32.2 g,三乙胺42.7 mL和吡咯烷(13d)7.3 g(62 mmol),反应1 h。加水100 mL,用乙酸乙酯(3×200 mL)萃取,合并有机相,浓缩后经硅胶柱层析(洗脱剂:A=5 ∶1)纯化得黄色固体苄基3-【5-{[(叔丁氧基羰基)胺基]甲基}-4-(吡咯烷-1-羰基)噁唑-2-基】吡咯烷-1-甲酸叔丁酯(14d),直接投入下步反应。
在反应釜中加入所得到的全部14d, 20%Pd(OH)2/C 6.27 g和MeOH 200 mL,通入氢气(0.3 MPa),于室温反应2 h。过滤,滤液浓缩得黄色固体[2-(吡咯烷-3-基)-4-(吡咯烷-1-羰基)噁唑-5-基]甲基胺基甲酸叔丁酯(15d)。
以13e替代13d,用类似的方法合成黄色固体(4-氨基甲酰基-2-吡咯烷-噁唑)- 5-甲基胺基甲酸叔丁酯(15e)。
15d: 收率75%;1H NMRδ: 5.88(s, 1H), 4.53(d,J=6.4 Hz, 2H), 3.91(t,J=6.4 Hz, 2H), 3.60(t,J=6.8 Hz, 2H), 3.42~3.37(m, 1H), 3.24~3.12(m, 3H), 3.05~2.98(m, 1H), 2.24~2.04(m, 2H), 1.95~1.87(m, 4H), 1.42(s, 9H); ESI-MSm/z: {[M+H]+}365.2。
15e: 收率81%;1H NMRδ: 6.96(s, 1H), 5.94(s, 1H), 5.64(s, 1H), 4.57(d,J=6.4 Hz, 2H), 3.45~3.00(m, 5H), 2.16(m, 2H), 1.43(s, 9H); ESI-MSm/z: {[M+H]+}311.1。
(14) 18f~18i的合成(以18f为例)
在多口瓶中加入15a 22 g(67 mmol)和甲醇240 mL,搅拌下于0 ℃滴加37%甲醛(16f)溶液18.1 g(223 mmol),滴毕(10 min),于室温反应0.5 h。加入三乙酰氧基硼氢化钠28.0 g,反应1 h。加入饱和碳酸氢钠溶液50 mL,用乙酸乙酯(3×100 mL)萃取,合并有机相,用无水硫酸钠干燥得淡黄色黏稠液体[4-甲基胺基甲酰基-2-(1-甲基-吡咯烷)-5-甲基-噁唑]-胺基甲酸叔丁酯(17f)。加入甲醇100 mL,搅拌下于0 ℃滴加混合溶液[m(盐酸)/m(乙醚)=1/4]20 mL,滴毕,于室温反应0.5 h。反应液浓缩后得淡黄色固体18f 15.8 g。
以乙醛(16g)和丙酮(16h)替代16f,用类似的方法合成黄色固体5-胺基甲基-2-(1-乙基吡咯烷)-噁唑-N-甲基甲酰胺盐酸盐(18g)和白色固体5-胺基甲基-2-(1-异丙基-吡咯烷)-噁唑-N-甲基甲酰胺盐酸盐(18h)。
18f: 收率85%;1H NMRδ: 4.74(d,J=5.2 Hz, 2H), 3.46~3.38(m, 1H), 2.92(s, 3H), 2.85~2.74(m, 2H), 2.66~2.54(m, 2H), 2.33(s, 3H), 2.27~2.10(m, 2H); ESI-MSm/z: {[M+H]+}238.8。
18g: 收率77%;1H NMRδ: 4.41(s, 2H), 4.04~3.85(m, 2H), 3.75~3.72(m, 2H), 3.52~3.49(t,J=8.0 Hz, 1H), 3.32~3.30(m, 1H), 3.28~3.25(m, 2H), 2.82(s, 3H), 2.59~2.40(m, 2H) 1.31~1.29(t,J=7.2 Hz, 3H); ESI-MSm/z: {[M+H]+}253.4。
18h: 收率82%;1H NMRδ: 3.96(s, 2H), 3.49~3.40(m, 1H), 3.04(t,J=9.6 Hz, 1H), 2.81~2.72(m, 5H), 2.65~2.59(m, 1H), 2.43~2.34(m, 1H), 2.24~2.13(m, 2H), 1.85~0.99(m, 6H); ESI-MSm/z: {[M+H]+}266.9。
(15) 18i的合成
在多口瓶中加入15a 13 g(44 mmol),三乙胺8.3 g(77 mmol)和二氯甲烷120 mL,搅拌下于0 ℃滴加乙酰氯(16i)3.5 g(44 mmol),滴毕(5 min),于室温反应0.5 h。反应液依次用1 mol·L-1盐酸(20 mL),饱和碳酸氢钠溶液(20 mL)和氯化钠溶液(20 mL)洗涤,合并有机相,浓缩,残余物经硅胶柱层析(洗脱剂:A=10 ∶1)纯化得淡黄色黏稠液体[2-(1-乙酰基-吡咯烷)-4-甲基胺基甲酰基-噁唑-5-甲基]-胺基甲酸叔丁酯(17i)。加入甲醇100 mL,搅拌下于0 ℃滴加混合溶液[m(盐酸)/m(乙醚)=1/4]10 mL,滴毕,于室温反应1 h。反应液浓缩,真空干燥得白色固体2-(1-乙酰基-吡咯烷)-5-胺基甲基-噁唑-N-甲基甲酰胺盐酸盐(18i),收率91%;1H NMRδ: 4.40(s, 2H), 4.00~3.50(m, 5H), 2.80(s, 3H), 2.41~2.20(m, 2H), 2.10(s, 3H); ESI-MSm/z: {[M+H]+}267.1。
2结果与讨论
合成15a~15e时,偶联剂对其收率影响较大,结果见表1。由表1可见,TUTB为偶联剂,15a~15c收率明显高于PyBOP; PyBOP为偶联试剂,15d和15e收率高于TUTB。

表1 偶联剂对15a~15e收率的影响

表2 溶剂对18f~18i收率的影响
合成18f~18i时,溶剂对其收率有较大影响,结果见表2。由表2可见,以甲醇为溶剂,合成18f~18i,收率较高(77%~85%);以二氯甲烷为溶剂合成18i,收率较高(91%)。
参考文献
[1]张慧珍,周成合,耿蓉霞,等. 噁唑类化合物合成研究新进展[J].有机化学,2011,12(31):1963-1976.
[2]Hartmann T C, Keeley D L, Pollock K M,etal. One-and two-photon fluorescent polyhedral oligosilsesquioxane(POSS) nanosensor arrays for the remote detection of analysis in clouds, in solution,and on surfaces[J].Chemistry of Materials,2008,20(8):2829-2838.
[3]Perner R J, Koenig J R, DiDomenico S,etal. Synthesis and biological evaluation of 5-substituted and 4,5-disubstituted-2-arylamino oxazole TRPV1 antagonists[J].Bioorganic & Medicinal Chemistry Letters,2010,18(13):4821-4829.
[4]Li D R, Zhang D H, Sun C Y,etal. Total synthesis of phorboxazole B[J].Chemistry-A European Journal,2006,12(25):1185-1204.
[5]Skepper C K, Quach T, Molinski T F,etal. Total synthesis of enigmazole A from cinachyrella enigmatica:Bidirectional bond constructions with an ambident 2,4-disubstituted oxazole synthon[J].J Am Chem Soc,2010,132(30):10286-10292.
[6]常鹏,杨红伟,程广斌. 除草剂安全剂双苯噁唑酸的合成[J].精细化工中间体,2014,44(2):21-24.
[7]Barbachyn M R, Ford C W. Oxazolidinone structure-activity relationships leading to linezolid[J].Angew Chem Int Ed,2003,42(43):2010-2023.
[8]赵洪娟,张月琴. 维吉霉素类抗生素新化合物X-435-C的产生菌X-435的初步分类鉴定[J].中国抗生素杂志,2000,25(2):87-90.
[9]张俊松,徐鸣夏. 吡咯烷类生物碱新衍生物合成及解痉活性[J].中国药物化学杂志,1995,5(2):109-112.
[10]张杰,王强,徐文方,等. 吡咯烷类神经氨酸酶抑制剂的设计、合成与初步活性研究[J].中国药学杂志,2008,43(4):314-318.
Synthesis of Novel 2-(Pyrrolidin-3-yl)oxazole Derivatives
LI Cui-ping,MA Zhen-zhen,LIU Yan-ling,FAN Zhen,SUN Chun-ling,WANG Wei*
(Topfond Pharmaceutical Co., Ltd., Zhumadian 463000, China)
Abstract:According to the substructure-chaining method, five novel tert-butyl {[4-(methylcarbamoyl)-2-(pyrrolidin-3-yl)oxazol-5-yl]methyl}carbamate analogues(15a~15e) were prepared by twelve step reactions of esterification, condensation, cyclization, hydrolyzation, salification, etc, using trimethysilyl chloride and benzylamine as materials. Four novel 5-(aminomethyl)-N-methyl-2-(1-methylpyrrolidin-3-yl)oxazole-4-carboxamide hydrochloride derivatives were synthesized by the reaction of 15a with aldehyde(ketone) or acetylchloride. The structures were characterized by1H NMR and ESI-MS.
Keywords:substructure-chaining method; methylcarbamoyl; oxazole; pyrrolidine; synthesis
收稿日期:2015-05-11;
修订日期:2016-03-21
基金项目:河南省科技攻关项目(112102310066)
作者简介:李翠平(1969-),女,汉族,河南驻马店人,工程师,主要从事药物生产和研发工作。 E-mail: li01358@163.com通信联系人: 王伟,教授级高工, E-mail: 1367564068@qq.com
中图分类号:O626.2
文献标志码:A
DOI:10.15952/j.cnki.cjsc.1005-1511.2016.05.15193
