盐酸苯达莫司汀的合成
2016-06-12崔美姬谌志华王庆庆虞心红
殷 昕, 崔美姬, 江 淼, 谌志华, 王庆庆, 虞心红
(华东理工大学 药学院,上海 200237)
·制药技术·
盐酸苯达莫司汀的合成
殷昕, 崔美姬, 江淼, 谌志华, 王庆庆, 虞心红*
(华东理工大学 药学院,上海200237)
摘要:以2,4-二硝基氯苯为起始原料,依次经甲胺化,选择性还原,N-酰化,环合,催化氢化,N-二羟乙基化,O-甲磺酰化,氯置换,酸化水解和成盐等10步反应合成盐酸苯达莫司汀,其结构经1H NMR确证,总收率48.4%,纯度99.5%。
关键词:2,4-二硝基氯苯; 苯达莫司汀; 药物合成; 工艺改进
盐酸苯达莫司汀(1),商品名为Treanda(SDX-105),是一种烷化剂类抗肿瘤药物。1主要通过烷化作用与DNA交联,干扰DNA功能和合成。此外,1还可使DNA和蛋白,蛋白和蛋白之间产生交联,从而发挥抗肿瘤作用[1-2]。1作为单用或联合化疗药物,对何杰金氏淋巴瘤和非何杰金氏淋巴瘤的治疗反应率分别为61%~97%和41%~48%。1对浆细胞瘤(多发性骨髓瘤)、慢性淋巴细胞白血病和乳腺癌等恶性肿瘤也有较好的疗效[3]。
根据起始原料,1的合成路线可分为以下两大类。方法一[4-10]:以2,4-二硝基氯苯为原料,依次与甲胺发生甲胺化反应,用硫化钠选择性还原邻位硝基,与戊二酸酐发生N-酰化反应,在浓硫酸催化下环合反应制得中间体——[1-甲基-2-(4′-丁酸乙酯基)-5-硝基]-1H-苯并咪唑(A); A先经催化氢化将硝基还原为氨基,再与环氧乙烷发生N-双羟乙基化[4-6]反应或与氯乙酸发生N-双羧基乙基化反应后还原得N-双羟乙基化物(B)[7]; B先经氯置换反应将羟基转化为氯,再依次经水解和成盐反应合成1,总收率33.5%~43.0%。方法二[11-12]:以4-硝基邻苯二胺为原料,经戊二酸环合,酯化和碘甲烷甲基化反应制得A; A经与方法一类似的路线合成1,总收率21.7%~45.0%。方法二不仅涉及价贵剧毒的碘甲烷,而且在引入甲基时还可能产生异构体。
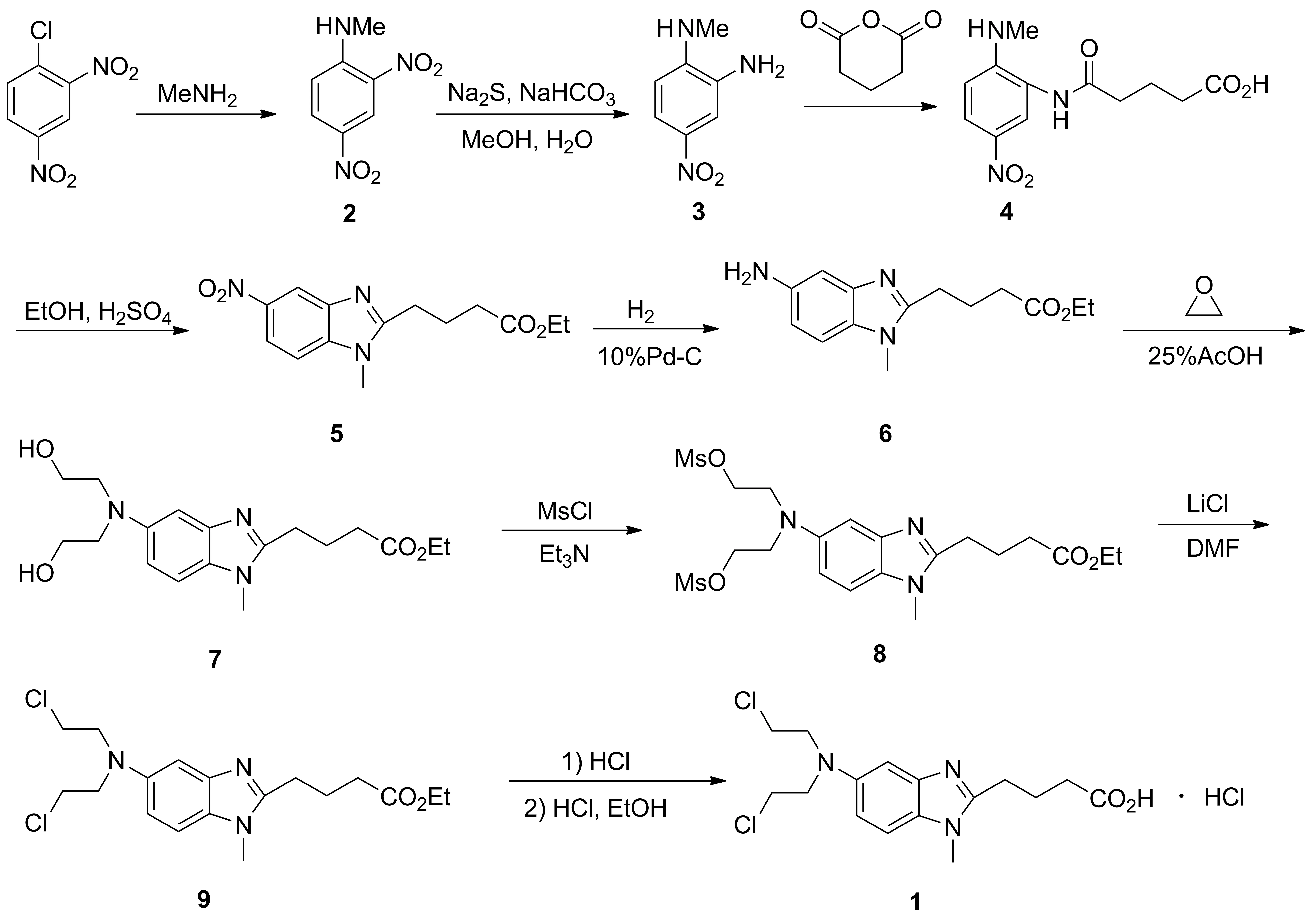
Scheme 1
本文在方法一的基础上进行了工艺改进。以2,4-二硝基氯苯为原料,依次经甲胺化,选择性还原,N-酰化,环合,催化氢化,N-二羟乙基化,O-甲磺酰化,氯置换,酸化水解和成盐等10步反应合成1(Scheme 1),其结构经1H NMR确证,总收率48.4%,纯度99.5%。该合成路线具有总收率较高,纯度较好,操作简便,条件温和等优点。
1实验部分
1.1仪器与试剂
WRS-1B型熔点仪(温度未校正);Bruker Avance-400型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Agilent 1100 Series型液相色谱仪。
所用试剂均为分析纯或化学纯。
1.2合成
(1)N-甲基-2,4-二硝基苯胺(2)的合成
0 ℃下,在反应瓶中依次加入33%甲胺醇溶液310 mL和2,4-二硝基氯苯40.5 g(0.20 mol),搅拌下于室温反应2 h。抽滤,滤饼于60 ℃真空干燥12 h得黄色粉末状结晶2 38.5 g,收率97.7%, m.p.177.8~179.1 ℃(收率96%, m.p.178~180 ℃[9]);1H NMRδ: 8.83(d,J=2.7 Hz, 1H, ArH), 8.26(dd,J=2.6 Hz, 9.6 Hz, 1H, ArH), 7.13(d,J=9.6 Hz, 1H, ArH), 3.33(s, 1H, NH), 3.06(d,J=5.0 Hz, 3H, CH3)。
(2) 2-氨基-4-硝基-N-甲基苯胺(3)的合成
在反应瓶中加入甲醇335 mL和2 7.9 g(0.04 mol),于50 ℃搅拌均匀;滴加九水合硫化钠36.5 g(0.15 mol)和碳酸氢钠12.8 g的水(120 mL)溶液,滴毕,回流反应30 min。趁热倒入800 mL冰水中,静置12 h;抽滤,滤饼用水(300 mL)洗涤,真空干燥24 h得紫红色晶体3 5.8 g,收率87.3%, m.p.177.1~178.2 ℃(收率88%, m.p.177~178 ℃[9]);1H NMRδ: 7.55(dd,J=1.9 Hz, 8.8 Hz, 1H, ArH), 7.41(d,J=2.2 Hz, 1H, ArH), 6.42(d,J=8.9 Hz, 1H, ArH), 6.12(d,J=4.2 Hz, 1H, NH), 5.08(s, 2H, NH2), 2.84(d,J=4.7 Hz, 3H, CH3)。
(3) 5-(2-甲氨基-5-硝基苯基)氨基-5-氧代戊酸(4)的合成
在三口瓶中加入3 16.7 g(0.10 mol),戊二酸酐14.5 g(0.10 mol)和二氯甲烷200 mL,回流反应3 h。冷却至室温,抽滤,滤饼用二氯甲烷(30 mL)洗涤,于60 ℃真空干燥12 h得淡黄色粉末4 26.6 g,收率94.7%, m.p.170.8~172.3 ℃(收率96%, m.p.170.5~171.5 ℃[9]);1H NMRδ: 9.20(s, 1H, OH), 7.99(dd,J=2.4 Hz, 9.1 Hz, 2H, ArH), 6.67(d,J=9.2 Hz, 1H, ArH), 3.34~3.93(m, 1H, NH), 2.85(d,J=4.7 Hz, 3H, CH3), 2.41(t,J=7.3 Hz, 2H, CH2), 2.30(t,J=7.0 Hz, 2H, CH2), 1.83~2.12(m, 2H, CH2)。
(4) [1-甲基-2-(4′-丁酸乙酯基)-5-硝基]-1H-苯并咪唑(5)的合成
在反应瓶中加入乙醇350 mL和4 65.0 g(0.23 mol),搅拌下滴加浓硫酸18.5 mL,回流反应2 h。冷却至室温,反应液倒入10%碳酸钠溶液(350 mL)中,静置析晶12 h;抽滤,滤饼用水(30 mL)洗涤,于60 ℃真空干燥12 h得类白色粉末5 61.0 g,收率91.4%, m.p.108.1~109.5 ℃(收率99%, m.p.109~110 ℃[9]);1H NMRδ: 8.50(s, 1H, ArH), 8.10(d,J=8.9 Hz, 1H, ArH), 7.29~7.58(m, 1H, ArH), 4.08(q,J=7.1 Hz, 2H, OCH2), 3.79(s, 3H, NCH3), 2.97(t,J=7.6 Hz, 2H, CH2), 2.50(t,J=6.9 Hz, 2H, CH2), 2.14~2.19(m, 2H, CH2), 1.22(t,J=7.1 Hz, 3H, CH3)。
(5) [1-甲基-2-(4′-丁酸乙酯基)-5-氨基]-1H-苯并咪唑(6)的合成
在反应瓶中加入甲醇600 mL和5 37.0 g(0.13 mol),搅拌使其溶解;加入10%钯碳2.0 g,常压下通入氢气,反应12 h。抽滤,滤液减压浓缩得粉红色固体6 32.1 g,收率96.8%, m.p.132.6~132.8 ℃(收率97.7%, m.p.130~132 ℃[8]);1H NMRδ: 7.03(d,J=8.0 Hz, 1H, ArH), 6.99(s, 1H, ArH), 6.65(dd,J=1.6 Hz, 8.4 Hz, 1H, ArH), 4.07(q,J=7.1 Hz, 2H, OCH2), 3.65(s, 2H, NH2), 3.63(s, 3H, NCH3), 2.87(t,J=7.6 Hz, 2H, CH2), 2.45(t,J=7.1 Hz, 2H, CH2), 2.14~2.32(m, 2H, CH2), 1.23(t,J=7.1 Hz, 3H, CH3)。
(6) [1-甲基-2-(4′-丁酸乙酯基)-5-双(2-羟乙基)氨基]-1H-苯并咪唑(7)的合成
0 ℃下,在反应瓶中依次加入25%冰醋酸30 mL, 6 5.2 g(20.00 mmol)和环氧乙烷6 mL(0.12 mol),搅拌下于室温反应12 h。用饱和碳酸钠溶液调至pH 7,用二氯甲烷(2×30 mL)萃取,合并萃取液,有机相用无水硫酸钠干燥,抽滤,滤液减压浓缩得淡棕色黏稠液体7 6.9 g,收率98.6%(96.6%[5]);1H NMRδ: 7.14(d,J=8.8 Hz, 1H, ArH), 7.08(s, 1H, ArH), 6.84(d,J=5.9 Hz, 1H, ArH), 4.12(q,J=7.1 Hz, 2H, OCH2), 3.82(d,J=3.9 Hz, 4H, NCH2), 3.69(s, 3H, NCH3), 3.55(d,J=4.4 Hz, 6H, HOCH2), 2.90(t,J=7.4 Hz, 2H, CH2), 2.46(t,J=6.9 Hz, 2H, CH2), 2.14~2.32(m, 2H, CH2), 1.25(t,J=6.9 Hz, 3H, CH3)。
(7) [1-甲基-2-(4′-丁酸乙酯基)-5-双(2-氯乙基)氨基]-1H-苯并咪唑(9)的合成
在三口瓶中加入7 5.2 g(15.00 mmol),二氯甲烷60 mL和甲磺酰氯3 mL(37.50 mmol),搅拌下于0 ℃滴加三乙胺5 mL,滴毕,反应4 h。抽滤,滤液用饱和碳酸氢钠溶液(2×10 mL)洗涤,用无水硫酸钠干燥,减压浓缩得黄色油状液体[1-甲基-2-(4′-丁酸乙酯基)-5-双(2-甲磺酰氧基乙基)氨基]-1H-苯并咪唑(8)8.3 g,未经纯化直接投入下一步反应。
在反应瓶中加入DMF 20 mL, 8 8.3 g(50.00 mmol)和无水氯化锂3.2 g(75.00 mmol),搅拌下于90 ℃反应3 h。减压浓缩,残余物加入饱和碳酸氢钠溶液(40 mL)中,搅拌30 min;抽滤,滤饼用水(2×10 mL)洗涤,于60 ℃真空干燥12 h得淡黄色固体9 5.2 g,收率90.0%(以7计), m.p.71.9~73.2 ℃;1H NMR δ: 7.24(d,J=8.6 Hz, 1H, ArH), 7.12(s, 1H, ArH), 6.82(d,J=8.5 Hz, 1H, ArH), 4.13(q,J=7.1 Hz, 2H, OCH2), 3.77(s, 4H, NCH2), 3.73(s, 3H, NCH3), 3.65(s, 4H, ClCH2), 2.99(d,J=24.4 Hz, 2H, CH2), 2.50(t,J=6.2 Hz, 2H, CH2), 2.19(d,J=10.9 Hz, 2H, CH2), 1.26(t,J=4.9 Hz, 3H, CH3)。
(8) 1的合成
在反应瓶中加入浓盐酸10 mL和9 3.9 g(10.00 mmol),搅拌下回流反应4 h。加入活性炭0.2 g脱色30 min;过滤,滤液减压浓缩,残余物用40%氢氧化钠溶液调至pH 4(冰浴冷却),析出白色晶体;过滤,滤饼用水(2×10 mL)洗涤,于60 ℃真空干燥12 h得白色粉末状结晶;用无水乙醇25 mL溶解,通入干燥氯化氢气体12 h,于0 ℃静置析晶12 h;抽滤,滤饼于35 ℃真空干燥12 h得白色结晶1 3.1 g,收率95%,纯度99.5%, m.p.149.1~150.8 ℃(m.p.148~151 ℃[10])。
由.5合成6时,采用钯碳进行催化还原,避免了由铁粉或雷尼镍还原产生的杂质及废料,使反应更加完全,收率更高。此外,钯碳可以回收使用。
由6合成7时,直接采用环氧乙烷进行N-双羟乙基化,与氯乙酸相比,不需还原羧基,可缩短反应步骤,提高收率。采用低毒、易处理的二氯甲烷代替氯仿作萃取剂,降低了成本。
由7合成9时,先将羟基转化为易于离去的甲磺酸酯,再进行氯置换,避免使用文献[4,6,7]方法中对设备腐蚀严重、对环境造成污染的氯化亚砜和三氯氧磷等氯化试剂,延长了设备使用寿命,减少了废气排放,有效提高了产物纯度。
由9合成1时,采用浓盐酸水解,活性炭脱色,氯化氢成盐的实验顺序,不需重结晶,缩短了反应时间,减少了浓盐酸用量,显著提高了1的纯度和收率。
参考文献
[1]陈祥峰,马俊杰. 苯达莫司汀的药理与临床研究[J].中国新药杂志,2011,20(8):665-668.
[2]王晓坤. 新型抗肿瘤药物-苯达莫司汀[J].齐鲁药事,2009,28(9):573-574.
[3]梁绍平,王华庆. 苯达莫司汀治疗淋巴瘤及白血病的研究进展[J].白血病·淋巴瘤,2013,22(8):456-465.
[4]陈磊,叶东,杨建楠,等. 盐酸苯达莫司汀的合成工艺改进[J].中国药师,2013,16(2):251-252.
[5]蔡瞻,姜志辉,赵明珠,等. 盐酸苯达莫司汀合成工艺的改进[J].药学实践杂志,2011,29(6):453-454.
[6]王先登, 刘立力, 张广明. 一种高纯度的盐酸苯达莫司汀的合成方法:CN 101 691 359A [P].2009.
[7]梁西周. 苯达莫司汀的合成优化[J].中国化工贸易,2013,6(6):288-289.
[8]高文磊,马帅,李少华,等. 盐酸苯达莫司汀的合成工艺研究[J].化学研究与应用,2014,26(10):1679-1682.
[9]高丽梅,汪燕翔,宋丹青. 盐酸苯达莫司汀的合成[J].中国新药杂志,2007,16(23):1960-1970.
[10]穆楠. 盐酸苯达莫司汀的合成[D].天津:河北工业大学,2013.
[11]Jian C, Katrin P. Discover of a novel efficient and scalable route to bendmustine hydrochloride:The API in treanda[J].Org Process Res Dev,2011,15(5):1063-1072.
[12]戴延凤,张胜菊,付敏,等. 盐酸苯达莫司汀的合成[J].化学试剂,2010,32(8):753-755.
Synthesis of Bendamustine Hydrochloride
YIN Xin,CUI Mei-ji,JIANG Miao,CHEN Zhi-hua,WANG Qing-qing,YU Xin-hong*
(School of Pharmacy, East China University of Science and Technology, Shanghai 200237, China)
Abstract:Bendamustine hydrochloride was synthesized by a ten-step reaction of amination, selective reduction, N-acylation, cyclization, catalytic hydrogenation, hydroxyethylation, O-sulfonylation, chlorination, hydrolysis and salification, using 2,4-dinitrochlorobenzen as starting material. The structure was confirmed by1H NMR. The total yield and purity were 48.4% and 99.5%, respectively.
Keywords:2,4-dinitrochlorobenzen; bendamustine; drug synthesis; process improvement
收稿日期:2015-05-04;
修订日期:2016-03-10
基金项目:国家自然科学基金资助项目(21476078, 20972051); 上海市科技支撑计划(12431900900, 12431900902)
作者简介:殷昕(1987-),男,汉族,江苏无锡人,硕士研究生,主要从事药物化学的研究。 E-mail: 493941813@qq.com通信联系人: 虞心红,教授,博士生导师, E-mail: xhyu@ecust.edu.cn
中图分类号:O626.15; O625.6
文献标志码:A
DOI:10.15952/j.cnki.cjsc.1005-1511.2016.05.15185
