《中华人民共和国药典(2015年版)》(四部)中微生物检验相关通则的增、修订情况介绍
2016-05-09冯震范一灵杨美成
冯震+范一灵+杨美成
摘 要 目的:介绍《中华人民共和国药典(2015年版)》(四部)中微生物检验相关通则的主要增、修订情况。方法:比较本版和前版药典相关内容的主要差异。结果:本版药典中微生物检验相关通则体现了新的检验理念,在检验技术、检验方法、环境设施、培养体系和质量管理等方面均进行了增、修订。结论:本版药典中微生物检验相关通则更趋完善,已与国际全面接轨。
关键词 《中华人民共和国药典(2015年版)》 微生物检验 通则 增、修订
中图分类号:R921.2 文献标识码:C 文章编号:1006-1533(2016)07-0011-05
An introduction to the revision of microbiological test in the general principle of the Pharmacopoeia of the Peoples Republic of China (2015 version) volume IV
FENG Zhen*, FAN Yiling, YANG Meicheng**
(Division of Antibiotics, Shanghai Institute for Food and Drug Control, Shanghai 201203, China)
ABSTRACT Objective: To systematically introduce major supplement and revision in the general principle of the Pharmacopoeia of the Peoples Republic of China (Chinese Pharmacopoeia) (2015 version) volume IV. Methods: The main differences between Chinese Pharmacopoeia (2010 version) and Chinese Pharmacopoeia (2015 version) were compared. Results: New ideas in microbiological test were established in Chinese Pharmacopoeia (2015 version), and supplement and updating amendment were carried out in testing technology and methods, environmental facilities, culture media and quality management. Conclusion: The general principle related to microbiological test in Chinese Pharmacopoeia (2015 version) is tending to be perfect and has achieved the international pharmaceutical standard.
KEY WORDS Pharmacopoeia of the Peoples Republic of China (2015 version); microbiological testing; general principle; revision
微生物污染是影响药品质量和安全性的主要因素之一。《中华人民共和国药典(2015年版)》(以下简称为“2015年版《中国药典》”)已自2015年12月1日起正式实施,其中的微生物检验相关通则实现了全药典的统一,形成了比较完备的药品微生物检验标准体系。2015年版《中国药典》(四部)中的微生物检验相关通则已与国际全面接轨,在多个方面与主要发达国家药典保持了协调一致,对药品中污染菌的控制、杀灭、检出、鉴定以及微生物检验的环境控制、实验室质量管理等均作出了更明确的要求和说明。2015年版《中国药典》(四部)中微生物检验相关通则的增、修订体现了近年来我国药品微生物检验领域的新理念、新方法和新技术,现将此部分的主要增、修订情况介绍如下。
1 微生物检验新理念
2015年版《中国药典》(四部)中微生物检验相关通则的总体要求体现了新的检验理念。由于药品中微生物污染的来源复杂且存在不均匀性,故本版药典规定无菌、微生物限度的检查结果为不符合规定时不得复试,仅在充分证明试验结果无效后方可重试;同时,强调不仅需要关注检验方法本身,还应重视菌种管理、控制菌鉴定、环境设施及性能、培养基质量控制和实验室生物安全性等多个方面,以保证每次检验结果的准确、可靠。因为有限数量的检验样品情况不能代表整批药品的情况,所以无菌、微生物限度等的检验结果并不能完全反映全部药品的实际微生物污染情况。鉴于此,2015年版《中国药典》微生物检验标准体系致力于推动药品微生物检验实验室从简单的终产品检验实验室向微生物风险调查分析型实验室方向发展。
2 微生物检验相关通则的基本情况
2015年版《中国药典》(四部)中的微生物检验相关通则较前版药典相关附录有较大的变化,其中新增了4则通则、修订了6则,仅3则通则未作修订。增、修订后,微生物检验相关通则总数达13则(表1)。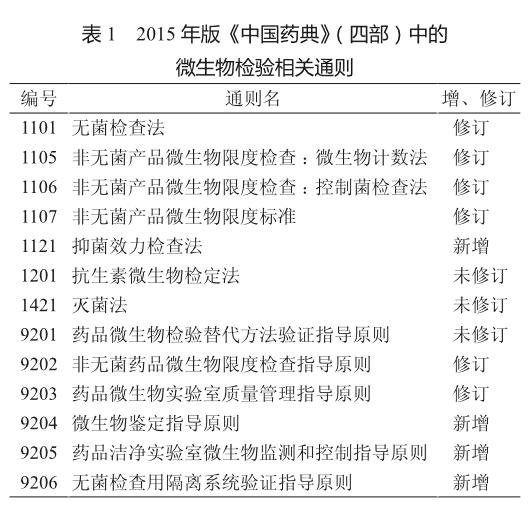
3 增、修订情况
下面简要介绍本版药典中微生物检验相关通则的增、修订情况,其中对药品检验工作中最为常用的无菌和微生物限度这两个检查项目所涉及的通则进行相对较为详细的论述。
3.1 无菌检查法
我国药典中的无菌检查法自1953年引入直接接种法至今已有63年的历史,将薄膜过滤法明确为无菌检查的首选方法也已有15年的历史,无菌检查技术已趋成熟。2005年后,我国药典在不断完善检验方法要求的基础上,逐渐建立起一个可控、可追溯的无菌药品检验体系。2015年版《中国药典》中的无菌检查法在实验环境、培养体系和质量控制等很多方面作了重大的调整和修订。
3.1.1 实验环境
避免微生物污染是无菌检查的首要条件,本版药典首次与2010年版《药品生产质量管理规范》中的洁净环境要求相衔接,将实验环境的洁净度级别分为A、B、C、D 4个等级。本版药典在编号为“9203”的通则中给出了无菌检查环境的指导性要求,即应在B级背景下的A级单向流洁净区或隔离系统中进行实验,强化了无菌检查过程中对环境的动态洁净度控制要求。
3.1.2 培养体系
在本版药典前,我国药品微生物检验领域普遍将微生物分为细菌和真菌两类,其中细菌接种于硫乙醇酸盐流体培养基(fluid thioglycollate medium, FTM),真菌接种于改良的马丁培养基。事实上,许多细菌和真菌在上述两种培养基中都能很好地生长和繁殖,而FTM的独特之处在于它可提供一种厌氧培养条件。在本版药典中,微生物的分类更多地依据其对营养物质的需求(尤其是对氧气的需求),这种分类方法更为合理和严谨。在微生物分类方法变化的基础上,为了与国际接轨,本版药典还采用胰酪大豆胨液体培养基替代改良的马丁培养基,并在培养基、方法适用性试验和供试品检查等部分作了相应的调整。例如,在培养及适用性试验中,金黄色葡萄球菌、铜绿假单胞菌和生孢梭菌接种于FTM后置于30 ~ 35 ℃下培养,枯草芽孢杆菌、白假丝酵母和黑曲霉接种于胰酪大豆胨液体培养基后置于20 ~ 25 ℃下培养,均延长观察时间至5 d。
3.1.3 供试品检查内容
本版与前版药典均强调无菌检查法的合理性和有效性,其中本版药典采用“方法适用性”替代“方法验证”,表述更为准确。在检验量和检验数量的定义上,本版药典根据国际通行的方法,按剂型和用途作了重新编排。同时,将前版药典中有关生物制品的内容进行了整合。在实验操作上,也明确了生物制品需要接种于2份FTM并分别置于30 ~ 35 ℃和20 ~ 25 ℃下进行培养、观察,以降低漏检风险。
3.1.4 风险评估和偏差调查的要求
本版药典倡导和强调了无菌检查过程中的污染风险评估和结果偏差调查体系建设的重要性。要充分理解本版药典无菌检查法的变化,不仅需要研读编号为“1101”通则自身的变化,而且还要结合编号为“9200”的通则,从现代化、规范化和国际化的微生物实验室质量管理角度出发,全面提高检验结论的可信度,重新审视无菌检查全过程中的风险管理和质量控制体系,紧跟本版药典指导理念的变化。
3.2 微生物限度检查法
2015年版《中国药典》中的微生物限度检查部分由原药典各部相应附录整合而成,包含编号为“1105”、“1106”和“1107”的3则通则。
3.2.1 实验环境
本版药典在微生物限度检查部分中并未明确规定检验环境的要求,而是在编号为“9203”的通则中给出了具体指导意见,即微生物限度检查应在不低于D级背景下的B级单向流空气区域内进行。本版药典的这一要求较之前的实验环境要求没有实质性的区别,并还在编号为“9205”的通则中对实验环境菌监控的范围和频次给出了更详细、更具体的指导原则,增强了方法的可操作性。
3.2.2 培养体系
微生物限度检查计数用培养基由胰酪大豆胨琼脂培养基、沙氏葡萄糖琼脂培养基替代了营养琼脂培养基、玫瑰红钠琼脂培养基。控制菌检查用培养基中,增菌培养基由胰酪大豆胨液体培养基替代了胆盐乳糖培养基和营养肉汤培养基,并新增了沙氏葡萄糖液体培养基。大肠埃希菌检查用培养基新增了麦康凯肉汤培养基,沙门菌检查用培养基由RV沙门菌增菌液体培养基替代了四硫磺酸钠亮绿培养基。此外,新增了肠道增菌液体培养基、紫红胆盐葡萄糖琼脂培养基、木糖赖氨酸脱氧胆盐琼脂培养基等;前版药典收载的甘露醇氯化钠琼脂培养基、溴化十六烷基三甲铵琼脂培养基等,虽培养基的名称未变,但配方中的营养成分、指示剂用量和筛选性能均有所改变。
需氧菌总数检查时的培养温度没有变化,为30 ~ 35℃,培养时间为3 ~ 5 d;霉菌和酵母菌计数检查时的培养温度由23 ~ 28 ℃改为20 ~ 25 ℃,培养时间为5 ~ 7 d;控制菌检查中的部分增菌培养时间有所延长,其中大肠埃希菌的培养温度改为42 ~ 44 ℃。
3.2.3 检查方法
3.2.3.1 微生物计数法
微生物计数法中新增了最可能数法,作为平皿法和薄膜过滤法的有效补充。最可能数法的精密度和准确度不及平皿法和薄膜过滤法,且仅能在对供试品需氧菌总数没有适宜计数方法时使用,不适用于霉菌计数。
微生物计数方法更加严谨、准确,计数结果以指数方式表示,最大可接受限度值遵守2倍规则,符合微生物污染的离散型泊松分布规律,能更客观地反映受试品中的微生物污染水平。
3.2.3.2 控制菌检查法
控制菌检查方法基本没有变化,但大肠埃希菌检查中新增了二次增菌步骤,耐胆盐革兰阴性菌检查中新增了预培养步骤。对肠道菌群的检查,修订了检查的名称,另由耐胆盐革兰阴性菌替代大肠菌群,检查范围更大。
3.2.4 方法适用性试验
计数回收试验中需要对铜绿假单胞菌、金黄色葡萄球菌、枯草芽孢杆菌、白假丝酵母和黑曲霉进行适用性检查,其中白假丝酵母和黑曲霉需要同时以胰酪大豆胨琼脂培养基和沙氏葡萄糖琼脂培养基进行计数回收试验。试验菌加入方式也有变化,要求将菌液加入供试液中,且所加菌液的体积不超过供试液体积的1%,使每ml供试液或每张滤膜所滤过的供试液中的含菌量<100 CFU。试验菌加入方式的改变更能确认方法的有效性和对微生物的无毒性。
计算适用性试验结果时,要求中和剂或灭活剂对照组的菌落数与菌液对照组菌落数的比值在0.5 ~ 2间,改变了原回收率≥70%的规定,更客观、合理。
3.2.5 微生物限度标准
本版药典中的微生物限度标准由原药典各部相关附录整合而成,并作了统一规定和合理调整:基于药品的给药途径及对患者的潜在危害风险等级,对齿龈、皮肤给药制剂,新增了大肠埃希菌、金黄色葡萄球菌和铜绿假单胞菌的检查;对呼吸道给药制剂,新增了耐胆盐革兰阴性菌的检查;对阴道、尿道给药制剂中含有中药成分者,新增了梭菌的检查;对原(辅)料、中药提取物和中药饮片,新增了微生物限度标准。此外,还加强了对沙门菌的控制。
本版药典还允许根据风险评估结果增加现未收载的控制项目,对建立个性化的药品质量标准作了原则性的规定。例如,对原(辅)料及某些特定制剂,根据其特性、生产工艺和用途等,可能还需检查其他具有潜在危害的微生物;药品中若检出其他可能具有潜在危害的微生物,应从给药途径、药品特性、使用方法和用药人群等方面进行评估;微生物风险评估人员应经过微生物学和微生物数据分析等方面的专业知识培训等。
3.3 抑菌效力检查法
对本身不具有充分抗菌效力的药品,应根据制剂(尤其是多剂量包装的制剂)特性添加适宜的抑菌剂,以防止制剂在正常贮藏或使用过程中可能发生的微生物污染和繁殖、从而使制剂变质而对用药者造成危害。所有抑菌剂都具有一定的毒性,故添加的抑菌剂的量应为最低有效量。同时,抑菌剂的抑菌效力在制剂贮藏过程中可能因受到药品成分或包装容器等因素的影响而发生变化,因此应验证制剂的抑菌效力不会在效期内因贮藏条件而降低。
2015年版《中国药典》将前版药典的“抑菌效力检查指导原则”修订为“抑菌效力检查法”,即由建议性要求变为了强制性控制要求,属重大改变。抑菌效力检查法用于检测灭菌及非灭菌制剂的抑菌活性,以评价最终药品的抑菌效力;也可用于生产企业,以指导和确定在研制剂中的抑菌剂浓度。该检查项目是制剂开发和留样稳定性考察时的重要检测项目,可保证制剂中的抑菌剂为最低有效量,且在制剂效期内抑菌有效。
3.4 药品微生物实验室质量管理指导原则
药品微生物检验受很多因素的影响,其中任何因素出现偏差都可能导致错误的检验结果。因此,实验室管理人员和检验者应了解所有影响检验结果准确性的环节和因素,进行严格的全面质量管理及过程控制,并采用适宜的质量控制方式予以监控。编号为“9203”的通则涵盖了以下几个方面的内容:人员、培养基、试剂、菌种、环境、设备、样品、检验方法、污染废弃物处理、检测结果质量保证、检测过程质量控制、实验记录、结果的判断、检测报告和文件等。
需要注意的是,药品微生物检验实验室应符合国家对生物安全的相关要求,尤其是对病原微生物的分离、鉴定应在二级或以上生物安全实验室中进行。
药品微生物检验实验室在建立、维持和改进管理体系时,宜将本指导原则与《药品生产质量管理规范》、《检测和校准实验室能力的通用要求》和《实验室资质认定评审准则》等相关规定一起作为管理体系的依据,以加强对实验室的专业、系统和具有针对性的质量管理。
3.5 微生物鉴定指导原则
本指导原则为非无菌药品微生物限度控制菌检查中疑似菌的鉴定以及制药原(辅)料、用水、中间体、终产品和环境中检出的微生物鉴定提供了指导,并明确指出微生物的鉴定以现行版的《伯杰氏系统细菌学手册》的鉴定结果为准。
本指导原则包括微生物的鉴定程序、方法确认和系统发育的相关内容以及溯源分析。除传统的表型鉴定技术外,还引入了基因型鉴定技术,如核酸测序、分子杂交、多位点序列分型和核糖体分型等。本指导原则还给出了微生物鉴定确认试验的方法及其指标,指标包括准确性、专属性、重现性、灵敏度、阳性预测值和阴性预测值,其中最重要的是准确性和重现性。
本指导原则是本版药典的新增内容,具有突破性的意义,将在通过对污染微生物进行鉴定、分型以确认污染来源,从而改进生产工艺、提高药品安全性,以及完善微生物实验室管理、保障检验结果准确性等方面发挥重要作用。
3.6 药品洁净实验室微生物监测和控制指导原则
本指导原则用于指导药品微生物检验用的洁净室等受控环境微生物污染情况的监测和控制,以使受控环境的微生物污染风险维持在可接受的水平内。本指导原则包括人员要求、初次使用的洁净室参数确认、微生物监测方法及频次、监测项目及标准、警戒限及纠偏限、数据分析及偏差处理、微生物鉴定和微生物控制等内容,对不同洁净级别区域内的浮游菌、沉降菌、接触碟和悬浮粒子等作出了明确规定。
3.7 无菌检查用隔离系统验证指导原则
本指导原则是为药典要求无菌的药品、生物制品、原(辅)料及其他产品无菌检查用隔离系统的验证提供指导的。
本版药典强调应尽可能保障无菌检查在最接近无菌的状态下进行,因此无菌检查用隔离系统的无菌保障能力必须得到保证。一般来说,ISO5(接近于动态A级)的污染率可以达到<1%,而无菌隔离器的污染率可以降低至<0.1%。隔离系统的验证是保障所需无菌环境的必要条件,在系统完成安装验证后应定期进行操作、完整性、灭菌、灭菌循环、内部洁净度和仪器、仪表的验证等。
4 结语
以上就2015年版《中国药典》(四部)中微生物检验相关通则的增、修订情况作了简要介绍。总之,本版药典紧跟国际先进药典的发展趋势,在微生物检验相关通则方面与国际通用标准相衔接,实现了自我革新和突破,完成了从注重检验结果到关注检验过程的重大转变。本版药典的实施有益于更好地提高我们的微生物检验相关技术能力和管理水平,从而加强药品的质量控制,保障我国药品的安全性和有效性。
