基于迁移学习的蛋白质交互关系抽取
2016-05-04李丽双黄德根周惠巍
李丽双,郭 瑞,黄德根,周惠巍
(大连理工大学 计算机学院,辽宁 大连 116023)
基于迁移学习的蛋白质交互关系抽取
李丽双,郭 瑞,黄德根,周惠巍
(大连理工大学 计算机学院,辽宁 大连 116023)
作为生物医学信息抽取领域的重要分支,蛋白质交互关系(Protein-Protein Interaction,PPI)抽取具有重要的研究意义。目前的研究大多采用统计机器学习方法,需要大规模标注语料进行训练。训练语料过少,会降低关系抽取系统的性能,而人工标注语料需要耗费巨大的成本。该文采用迁移学习的方法,用大量已标注的源领域(其它领域)语料来辅助少量标注的目标领域语料(本领域)进行蛋白质交互关系抽取。但是,不同领域的数据分布存在差异,容易导致负迁移,该文借助实例的相对分布来调整权重,避免了负迁移的发生。在公共语料库AIMed上实验,两种迁移学习方法获得了明显优于基准算法的性能;同样方法在语料库IEPA上实验时,TrAdaboost算法发生了负迁移,而改进的DisTrAdaboost算法仍保持良好迁移效果。
蛋白质交互关系抽取;迁移学习;负迁移
1 引言
伴随着信息数字化和生物医学领域的快速发展,生物医学文献呈爆发式增长,使得提取文献中的信息困难重重。生物医学文本挖掘应运而生。作为其中的重要分支之一,蛋白质交互关系(PPI)抽取技术具有很高的应用价值,尤其是对蛋白质知识网络的建立、本体的构建等具有重要意义。
目前用于蛋白质交互关系抽取的方法可以分为三种,基于词共现的方法,基于模式匹配的方法和基于统计机器学习的方法。统计机器学习方法是主流的方法,取得了较好的结果,它可分为基于特征向量和基于核函数[1]的方法以及组合方法[2],组合方法具有比单一方法更好的性能。
虽然基于统计机器学习的方法在蛋白质交互关系抽取领域得到了较好的应用,但是当训练语料极其匮乏时,抽取效果就会大大降低。人工标注PPI数据库需要耗费大量的人力和物力。从另一个角度讲,不能很好地利用大量的、已标注的语料,亦是对资源的浪费。例如在不同疾病的蛋白质交互关系的研究中,我们可以借助其他疾病领域的已标注数据集来提高本领域的抽取效果。
利用其他领域已标注信息,解决本领域训练数据不足问题,当下有两种思路:领域适应和迁移学习。其中,领域适应在人脸识别[3],情感分类[4],机器翻译[5]和中文分词[6]等领域得到很好的应用,也有研究者采用领域适应的方法探讨了PPI抽取,如文献[7]讨论了AIMed,IEPA,LLL,HPRD50和BioInfer五个语料在大小和物种方面的差异,提出一种统一的标注方法,并采用该方法处理五种语料,对结果进行定量分析,整合出更大的、物种更多的语料,用统一标注的其他领域语料辅助训练,在五个语料上测试的精确率、召回率、F值均有明显提高。文献[8]从上述五个语料出发,采用领域适应方法SVM-CW,同时对多个语料学习,调整源领域语料的权重,选取合适的惩罚因子,在五个语料上取得了较好的效果。但是文献[7]和文献[8]主要讨论领域间的兼容性,未对目标训练语料不足做出分析,使得辅助训练语料占全部训练语料比例较小,对目标训练语料的需求仍然较大,不能大幅减少标注目标语料的成本。
迁移学习是机器学习领域的一个新方向,是在不同但相似的领域、任务之间进行知识的迁移,在Web文本数据挖掘[9]、文本分类[10-11]、双语句子对齐[12],取得了很好的效果。但研究者很少将迁移学习应用到PPI抽取中。本文尝试采用迁移学习思想探讨因标注语料不足而导致PPI抽取性能较差的问题。但是领域间数据分布会存在差异,如不同物种间的生理特征,基因及蛋白质等存在显著不同,差异较大时容易导致负迁移[13],迁移学习反而降低了普通分类器的学习效果。针对上述问题,本文提出一种改进的DisTrAdaboost算法,通过调整其他领域已标注数据集的实例权重的方法来避免负迁移,即用实例的相对分布来初始化权重,增加源数据中分布近似于目标数据集的实例的权重,进而平滑学习过程,降低领域差异对迁移学习的影响,避免负迁移。
在蛋白质交互关系抽取任务上,我们首先把AIMed作为目标语料,选取IEPA和HPRD50作为辅助训练语料(源语料),分别采用TrAdaboost[14]算法和改进的迁移学习算法DisTrAdaboost进行实验。结果表明,两种迁移学习方法均获得了较好的抽取效果,并且没有发生负迁移。然后IEPA作为目标语料进行了相同的实验,以HPRD50作为源语料时,TrAdaboost算法发生了负迁移,而改进的DisTrAdaboost算法依然取得了很好的迁移效果,没发生负迁移。此外,在文本分类任务的公共评测语料上,我们验证了DisTrAdaboost算法的收敛性,得出DisTrAdaboost算法收敛速度明显快于TrAdaboost算法,分类效果也优于TrAdaboost算法。
2 改进的DisTrAdaboost算法
为使问题描述更清晰,我们给出以下定义:
定义2.1(基本符号):
X:样例空间,也指需要被分类的输入数据。
Y={-1,+1}: 类空间。
c(x): 样本x∈X的类标,且c(x)∈Y。
定义2.2(数据集):
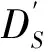
图1给出了TrAdaboost算法的描述。
TrAdaboost算法继承了Adaboost算法良好的收敛性,并且训练集中实例的初始权重不影响其收敛性。但是,TrAdaboost算法初始设定源领域数据集实例权重相等,使得迁移效果严重受到领域差异的影响,未能有效避免负迁移的发生。下面讨论数据分布对迁移学习的影响,并采用实例的分布来初始化源数据集实例的权重,降低分布差异对迁移学习的影响。
基于实例的迁移学习有以下假设:
(3)P(XS)≠P(XY)
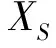

图1 TrAdaboost算法描述
迁移学习的目标就是,用源数据集DS辅助训练,在目标数据集DT上得到观测值hT,使得观测值尽可能接近目标值,即损失函数最小。为达到上述目标,Zadrozny[15]对损失函数l(x,y,θ)做出如下推导:
(1)
针对源数据集和目标数据集的分布差异较大问题,本文提出了一种DisTrAdaboost算法进行改进。首先,我们给出相对分布δ(x)的定义:
(2)
即,实例x在目标数据集的分布与在源数据集的分布之比。本文使用KL距离(见4.1节)来计算分布PT(x)和PS(x)。为使问题更具一般性,我们在式(1)中加入惩罚函数,求解参数θ*的过程就变为:
(3)
其中λ是正则化系数,Ω(θ)是正则化项。对于给定分类算法,损失函数l(x,y,θ)是固定值,求解θ*就转化为求解δ(xi)最小值问题。由此,我们可以看出改进后的目标领域损失函数小于原先的目标领域损失函数:
(4)
同理,不难得出,改进后的源领域损失函数小于之前的源领域损失函数:
(5)
本文采用最直观的做法,评价源领域数据集中每个实例Si的相对分布δ(xi)。δ(xi)取值越大,初始训练权重越低。
最终,改进后的DisTrAdaboost算法初始权重向量修改如式(6)所示。
(6)
TrAdaboost算法劣势在于,收敛速度较慢。当领域差异较大时,容易导致负迁移。考虑到目标领域训练数据和目标领域测试数据分布相同,训练损失较小,本文主要对源领域数据训练损失的迭代收敛速度进行讨论,如式(7)所示。
(7)
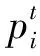
根据定义,DisTrAdaboost算法初始的平均加权训练损失为:
(8)
同理,TrAdaboost算法初始的平均加权训练损失为:
(9)
根据式(5)可以得出, DisTrAdaboost算法初始的平均加权训练损失小于TrAdaboost算法初始的平均加权训练损失,更快地到达收敛,从而更加有效地避免负迁移。
3 特征抽取
3.1 词特征
本文中运用了丰富的词特征,这些词特征包括两个蛋白质名字中的词,两个蛋白质之间的词,蛋白质周围的词,以及表示两个蛋白质交互关系的交互词。
(1) 两个蛋白质名字中的词(ProName)
指出现在两个蛋白质名字中的所有单词。
(2) 两个蛋白质之间的词(Betwords)
指位于两个蛋白质之间的所有词。如果两个蛋白质之间没有别的单词,那么该特征被设置为NULL。
(3) 两个蛋白质周围的词(Surrounding Words)
指两个蛋白质前后的词。本文选取交互关系对中第一个蛋白质的前面五个词和第二个蛋白质的后面五个词。
(4) 交互词特征(Keyword)
所谓交互词指的是能表示两个蛋白质之间交互关系的词(如:regulate, interact, modulate 等)。如果在两个蛋白质之间或者周围有一个交互词,则把这个词作为交互词特征。如果在一个句子中有两个或两个以上的交互词,则选择离两个蛋白质最近的那个词作为交互词;如果在一个句子没有表示两个蛋白质交互关系的词,则把该特征设置为NULL。
3.2 两个蛋白质之间的距离特征
在一个句子中,如果两个蛋白质离的越近,那么它们之间具有交互关系的可能性也就越大。因此蛋白质之间的距离可以作为判断两个蛋白质是否具有交互关系的一个因素。本文使用两种距离特征。
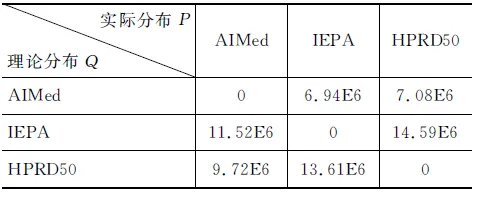
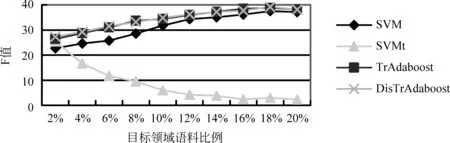
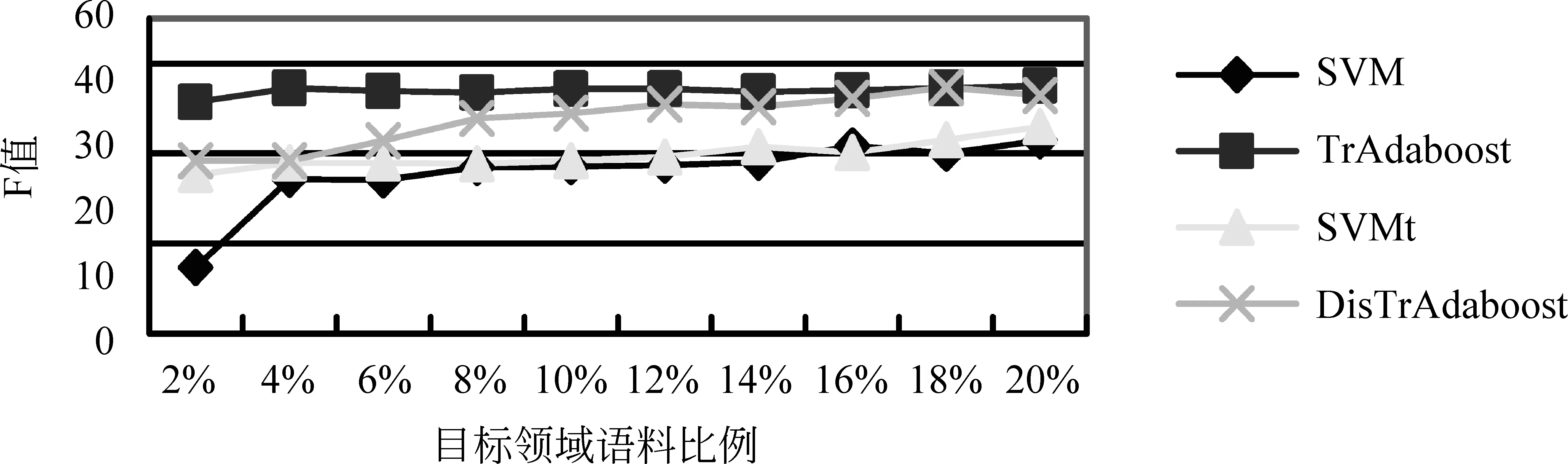
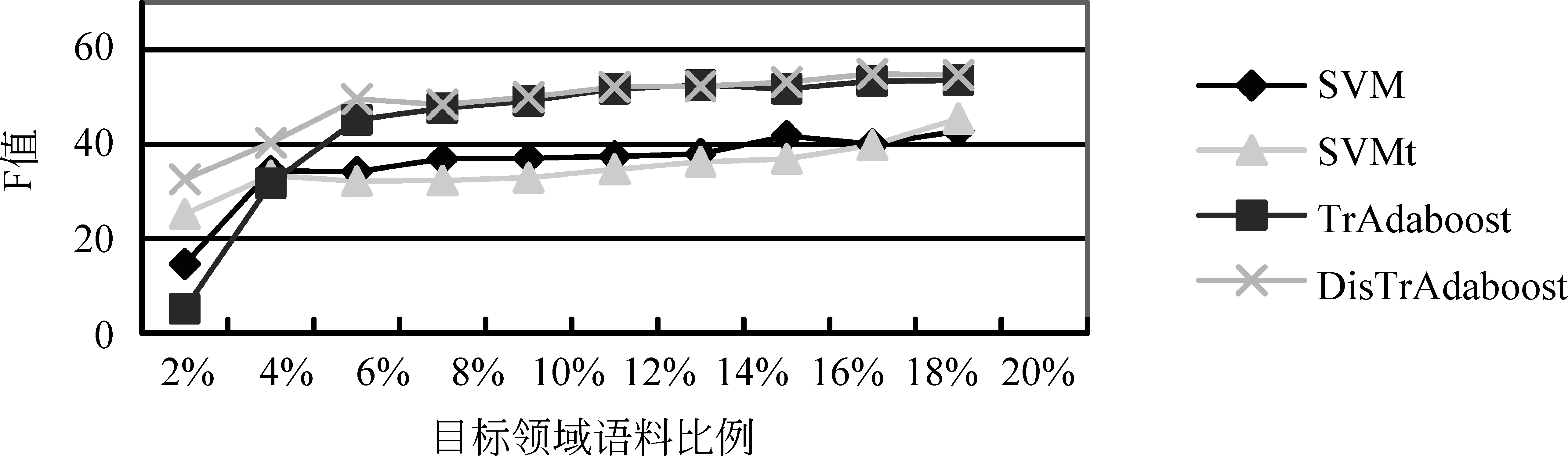
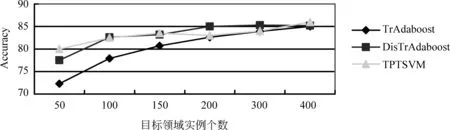
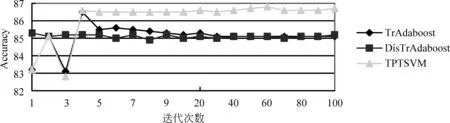
(1) TwoProDis: 指两个蛋白质之间其他单词的个数。如果TwoProDis≤3,则把该特征值设为1;如果3 (2) ProNumDis: 两个蛋白质之间是否具有交互关系,除了受两个蛋白质之间单词的个数影响外,还与两个蛋白质之间是否有别的蛋白质有关。因此这种情形也被考虑进去,并称之为ProNumDis 特征。如果两个蛋白质之间没有其它的蛋白质,那么该特征的特征值被设为0,如果两个蛋白质之间有其它蛋白质,那么该特征的特征值为两个蛋白质之间其它蛋白质的个数。 本文使用两种语料作为目标语料进行实验:AIMed语料和IEPA语料。前者来自Medline数据库中的225篇摘要,有1 000对正例(有交互关系),3 500对负例(无交互关系)。后者摘自PubMed数据库中的303篇摘要,其中正例336对,负例336对。另外,还选取了HPRD50语料作为源领域语料。HPRD50有163条正例和270条负例。本文使用F值对该实验进行评价。它的定义如式(10)所示。 (10) 其中,TP(true positive)表示所有正例中判断正确的样本数。FN(false negative)代表负例中判断错误的样本数,即分类器把正例判断成负例的个数。FP(false positive)代表正例中判断错误的样本数,即分类器把负例判断成正例的样本个数。其中,F值为准确率和召回率的调和函数值。实验均采用五倍交叉方法。 4.1 可迁移性评价 为了更好刻画语料间分布差异,我们引入KL距离[16]。计算如式(11)所示。 (11) 式(11)中P和Q是两个概率分布,D(P||Q)是对P和Q差别的非对称性的度量,又名KL散度。KL散度是用来度量使用基于Q的编码来编码来自P的样本平均所需的额外的比特个数。典型情况下,P表示数据的真实分布,Q表示数据的理论分布,模型分布,或P的近似分布。且KL散度不具有对称性: 从分布P到Q的距离(或度量)通常并不等于从Q到P的距离(或度量)。 本文通过计算单词x在数据集P的出现频率作为P(x),同理可得Q(x)。 表1给出了三个数据集间的KL距离结果。因HPRD50数据集过小,本文未将其选作目标领域数据集。其它KL距离结果存在如下关系: (12) 从结果可以看出,数据集AIMed到IEPA的距离远小于数据集HPRD50到IEPA的距离。 表1 AIMed,IEPA,HPRD50语料间的KL距离 4.2 AIMed作为目标领域时的迁移学习 将IEPA和HPRD50作为源领域数据集,随机选取AIMed语料的2%,4%,6%,8%,10%,12%,14%,16%,18%,20%作为目标领域训练集。将两种迁移学习算法与两个基准算法进行比较,分别是:(1)SVM,即简单地用目标领域语料训练;(2)SVMt,即合并源领域语料和目标领域语料训练。结果如图2和图3所示。 图2 IEPA作为源领域时,四种算法在AIMed上F值对比 图3 HPRD50作为源领域时,四种算法在AIMed上F值对比 从图2和图3中可以看出:首先,两种迁移学习算法曲线都一直高于SVM曲线,说明IEPA或HPRD50作为源语料时,迁移学习效果优于普通SVM;其次,两种算法曲线也一直高于SVMt曲线,说明都能够通过调整源数据集实例的权重,选取合适实例辅助训练,其效果优于直接在合并数据集上训练;最后,两种算法都未发生负迁移,说明迁移学习在该数据集上取得了较好的效果。 然而,对比两种迁移学习曲线,我们发现,在目标语料较少(2%~4%)时,算法DisTrAdaboost曲线高于TrAdaboost曲线,说明DisTrAdaboost算法通过加速收敛,从而更好地适应目标领域语料的不足;当目标语料达到一定程度后(≥8%),两个曲线基本重合,原因是随着目标领域语料的增多,TrAdaboost算法也取得了较好的收敛效果,DisTrAdaboost算法加速收敛的效果不再明显。 4.3 IEPA作为目标领域的结果对比 与上节实验类似,将两种算法与两个基准算法SVM和SVMt进行比较。结果如图4和图5所示。 图4 AIMed作为源领域时,四种算法在IEPA上F值对比 图5 HPRD50作为源领域时,四种算法在IEPA上F值对比 图4中,两种迁移学习曲线都高于SVM曲线和SVMt曲线,然而改进的DisTrAdaboost算法曲线低于TrAdaboost曲线。原因分析如下,4.1节,我们得出数据集AIMed到IEPA的KL距离较小,TrAdaboost算法迁移效果较好,此时,DisTrAdaboost算法对源数据集AIMed的数据分布进行调整,过滤掉了原本符合辅助训练条件的实例,降低了性能。 图5中,迁移学习曲线一直高于SVM曲线和SVMt曲线。由此可以得出类似图2的结论:以AIMed作为源语料,IEPA作为目标语料时,迁移学习可以明显提高PPI抽取性能。此外,由图5可以看出,在目标语料比例为2%和4%时,TrAdaboost算法曲线低于SVM和SVMt,说明此时产生了负迁移;而改进的DisTrAdaboost算法曲线仍一直高于SVM曲线和SVMt曲线,说明用相对分布β(xi)来初始化每个实例的权重,可以降低领域差异对迁移学习的影响,弥补目标训练语料过少导致基本分类器效果差的不足,避免负迁移。 4.4 与相关工作比较 为验证算法DisTradaboost算法的有效性,本文还在文本分类语料20newsgroups*http://qwone.com/~jason/20Newsgroups上进行了实验。该语料共分为七个大类,每个大类分为若干子类。本文选取以下两个大类作为分类任务:sci和talk。目标领域设为sci.space和talk.religion.misc,共2 315条实例。源领域则由与目标领域语料同属一个大类下的其他子类构成,包括sci.crypt,sci.electronic,sci.med,talk.politics.gunstalk.politics.mideast和talk.politics.misc,共4 880条实例。实验用精确率进行评价,定义如式(13)所示。 (13) 其中,True表示分类正确的实例个数,False表示分类错误的实例个数。 本文还将DisTrAdaboost算法和TPTSVM算法[11]的效果,进行了对比。其中,TPTSVM[11]结合了迁移学习和半监督学习的思想,利用了大量未标注数据辅助训练,效果好于TrAdaboost算法。 图6中,TPTSVM算法和DisTrAdaboost算法曲线都高于TrAdaboost曲线,说明前者利用大量无标记语料,后者通过加速收敛,都能改进TrAdaboost算法。对比TPTSVM算法曲线和DisTrAdaboost算法曲线,我们发现在目标领域训练语料较少(≤50)时,TPTSVM效果最好,说明该算法能够正确地标注目标领域的未标注语料,从而辅助训练,学习效果更好;在目标领域训练语料达到一定规模(≥200)后,半监督学习影响下降,而DisTrAdaboost算法能更好地使用源领域辅助训练数据,加速收敛。 图6 迁移学习算法在文本分类语料上的精确率对比 图7中,我们对比了TrAdaboost算法,DisTrAdaboost算法和TPTSVM算法在不同迭代次数时的学习曲线。其中,目标领域实例个数全部设为400。从学习曲线中,我们可以发现:三种算法最终都能达到收敛,而DisTrAdaboost算法初始收敛速度更快。 图7 三种迁移学习算法的学习曲线 4.5 语料分布对迁移学习效果影响分析 表1中,给出了上述三个数据集的KL距离的大小关系(式(12))。简单起见,仅对KL距离差异最大的两对数据集进行讨论。图4中,AIMed作为源数据集,IEPA作为目标数据集,KL距离最小,TrAdaboost算法曲线表现最好,一直高于另外三种算法曲线,说明用源数据集表示目标数据集的KL距离较小时,迁移学习表现良好,不必考虑负迁移;而在图5中,HPRD50作为源数据集,IEPA作为目标数据集,KL距离D(PIEPA‖QHprd50)较大,TrAdaboost在目标数据较少(仅占2%和4%)时,效果不如SVMt和SVM,而改进的DisTrAdaboost算法此时表现最好,说明KL距离较大容易导致负迁移,通过调整语料的初始权重,可以降低语料分布差异对迁移学习的影响。 在蛋白质交互关系抽取领域,训练语料不足时,传统机器学习方法分类效果很差,为此我们引入迁移学习。以TrAdaboost算法作为迁移实现,SVM和SVMt作为参照实验,实验结果表明,迁移学习借助源领域语料,只需标注小部分目标领域语料,就可以达到一定的抽取性能。对比发现,当目标语料较少并且源语料和目标语料分布相近时,迁移效果远好于SVM,随着训练语料的增加,SVM逐渐逼近迁移学习。同时,针对负迁移问题,本文对TrAdaboost算法进行了改进,用实例的相对分布来初始化权重向量,实验结果表明,通过对源数据集中实例进行预先评价,利用实例在源数据集和目标数据集的分布,增加与目标数据集分布相似的实例的权重,降低与目标数据集分布相异的实例的权重,可以有效避免负迁移。并且在文本分类任务上,改进的DisTrAdaboost算法的收敛速度明显快于TrAdaboost算法,其分类效果也优于TrAdaboost算法。 本文工作对于近似领域知识的迁移具有借鉴作用,如不同疾病(肝病和肺病)的蛋白质交互关系抽取的研究。这些领域的数据分布不同但近似,对于缺少足够训练样本的目标领域,充分利用已有标记的源领域数据资源,采用迁移学习方法可以提升目标领域的知识挖掘性能。 [1]QianLH,ZhouGD.Dependency-directedtreekernel-basedprotein-proteininteractionextractionfrombiomedicalliterature[C]//Proceedingsofthe5thInternationalJointConferenceonNaturalLanguageProcessing,Thailand,2011: 10-19. [2] 李丽双,刘洋,黄德根. 基于组合核的蛋白质交互关系抽取[J]. 中文信息学报, 2013, 27(1): 86-92. [3] 陶剑文,王士同. 多核局部领域适应学习[J]. 软件学报,2012, 23(9):2297-2310 [4]LiSS,XueYX,WangZQandZhouGD.Activelearningforcross-domainsentimentclassification[C]//ProceedingsoftheTwenty-ThirdInternationalJointConferenceonArtificialIntelligence.Beijing,China, 2013: 2127-2133. [5] 苏晨,张玉洁,郭振,徐金安. 适用于特定领域机器翻译的汉语分词方法[J]. 中文信息学报,2013,27(05):184-190. [6] 孟凡东,徐金安,姜文斌,刘群. 异种语料融合方法:基于统计的中文词法分析应用[J]. 中文信息学报,2012, 26(2):3-7,12. [7]SampoP,AnttiA,JuhoH,etal.Comparativeanalysisoffiveprotein-proteininteractioncorpora[J].BMCBioinformatics,2008, 9:S6. [8]MiwaM,SaetreR,MiyaoY,etal.Arichfeaturevectorforprotein-proteininteractionextractionfrommultiplecorpora[C]//ProceedingsoftheAssociationforComputationalLinguistics,Singapore:WorldScientificPublishingCoPteLtd. 2009: 121-130. [9]WeiFM,ZhangJP,ChuY,etal.FSFP:TransferLearningFromLongTextstotheShort[J].AppliedMathematics&InformationSciences, 2014, 8(4): 2033-2040. [10]YangP,GaoW,TanQ,etal.Alink-bridgedtopicmodelforcross-domaindocumentclassification[J].InformationProcessing&Management, 2013, 49(6): 1181-1193. [11]ZhouH,ZhangY,HuangD,etal.Semi-supervisedLearningwithTransferLearning[J].ChineseComputationalLinguisticsandNaturalLanguageProcessingBasedonNaturallyAnnotatedBigData.SpringerBerlinHeidelberg, 2013: 109-119. [12] 陈相, 林鸿飞, 杨志豪. 基于高斯混合模型的生物医学领域双语句子对齐[J]. 中文信息学报, 2010, 24(4): 68-73. [13]PanSJ,YangQ.Asurveyontransferlearning[J].KnowledgeandDataEngineering. 2010, 22(10): 1345-1359. [14]DaiW,YangQ,XueGR,etal.Boostingfortransferlearning[C]//Proceedingsofthe24thInternationalConferenceonMachineLearning. 2007: 193-200. [15]ZadroznyB.Learningandevaluatingclassifiersundersampleselectionbias[C]//ProceedingsoftheTwenty-firstInternationalConferenceonMachineLearning. 2004: 114. [16]KullbackS,LeiblerRA.Oninformationandsufficiency[J].TheAnnalsofMathematicalStatistics, 1951, 22(1): 79-86. Protein-Protein Interaction Extraction Based on Transfer Learning LI Lishuang, GUO Rui, HUANG Degen, ZHOU Huiwei (School of Computer Science and Technology, Dalian University of Technology, Dalian, Liaoning 116023, China) As an important branch of biomedical information extraction, Protein-Protein Interaction (PPI) extraction has great research significance. Currently, research of PPI mainly focuses on traditional machine learning, which requires the use of large amounts of annotated corpus for training and makes it costly to label the new data. This paper employs Transfer Learning in extracting PPI with a small amount of labeled data of target domain (in-domain), drawing support from annotated data of source domain (out-of-domain). To avoid the negative transfer caused by large differences between the distributions of different domains, we adjust the weights of each instance from source domain, depending on its relative distribution. Experiments on the AIMed corpus and on IEPA corpus reveals the efficiency of our alogrithems. PPI; transfer learning; negative transfer 李丽双(1967—),博士,教授,主要研究领域为自然语言理解、信息抽取与机器翻译。E⁃mail:lils@dlut.edu.cn郭瑞(1990—),硕士研究生,主要研究领域为信息抽取。E⁃mail:guoruiaini1994@126.com黄德根(1965—),博士,教授,主要研究领域为自然语言理解与机器翻译。E⁃mail:huangdg@dlut.edu.cn 1003-0077(2016)02-0160-08 2013-12-10 定稿日期: 2014-11-28 国家自然科学基金(61173101, 61173100, 61272375) TP391 A4 实验及结果分析

5 总结

