新型Rh2(esp)2配体手性类似物的合成
2016-04-20黄泽傲卢崇道
黄泽傲, 卢崇道
(1. 中国科学院 新疆理化技术研究所,新疆 乌鲁木齐 830011; 2. 中国科学院大学,北京 100049)
·快递论文·
新型Rh2(esp)2配体手性类似物的合成
黄泽傲1,2, 卢崇道1*
(1. 中国科学院 新疆理化技术研究所,新疆 乌鲁木齐830011; 2. 中国科学院大学,北京100049)
摘要:以(R)-叔丁基亚磺酰胺为手性助剂,与1,3-苯二甲醛经缩合反应制得关键中间体——(Rs,Rs)-双叔丁基亚磺酰亚胺(7);锂化的羧酸酯与7经不对称加成反应合成了两个新型的Rh2(esp)2配体类似物——(Rs,Rs′,R,R′)-β-胺基羧酸酯和(Rs,Rs′,R,R′,R,R′)-α,β-氮杂环丙烷羧酸酯,产率分别为96%和65%,非对映选择性均大于20 ∶1。化合物的结构经1H NMR和(13)C NMR表征。
关键词:Rh2(esp)2; 双核铑(Ⅱ)催化剂; 叔丁基亚磺酰双亚胺; (Rs,Rs′,R,R′)-β-胺基羧酸酯; (Rs,Rs′,R,R′,R,R′)-α,β-氮杂环丙烷羧酸酯; 手性修饰; 合成; 非对映立体选择性
桥联双核铑(Ⅱ)催化剂是一类具有桨轮结构的化合物[1],其在催化C-H官能化等方面有广泛用途[2-6]。2004年,斯坦福大学Du Bois J等[7]首次合成了配体Rh2(esp)2(1, Chart 1)。在随后的多项研究中发现,1有着优越的催化C-H氨化的活性[7-11]。尽管如此,手性双核铑催化剂催化的不对称C-H氨化鲜见报道[12],人们更多地采用手性氮源作为控制C-H氨化立体选择性的手段[13]。因而,开发用于催化C-H不对称氨化的手性双核铑催化剂值得期待。鉴于1在催化C-H氨化方面所表现出的优越性,在1的结构中引入手性中心便成为设计新的手性催化剂的可能途径。到目前为止,基于配体α,α,α′,α′-四甲基-1,3-苯二丙酸(esp)(2)的手性修饰尚未见报道。我们曾对1的构效关系进行研究,从中发现间苯二甲基和羧基α-位的两个甲基是使催化剂稳定并具有优越催化性能的重要因素[10-11,14]。因此,开发1类手性催化剂,在保留苯二烷基结构和保持羧基α-位为季碳原子同时,需在2的A或B位置构建结构如3或4的配体前体羧酸酯(Chart 1)。
近年来,手性叔丁基亚磺酰胺在合成含氮化合物方面表现出了优越的立体选择性[15]。本课题组用其成功地发展了Brook重排介导的多组分反应[16-18]。本研究首次使用手性叔丁基亚磺酰胺对配体1进行手性修饰,逆合成分析如Chart 1所示。本文以(R)-叔丁基亚磺酰胺作为手性助剂,与1,3-苯二甲醛经缩合反应制得关键中间体——(Rs,Rs)-双叔丁基亚磺酰亚胺(7);锂化的羧酸酯与7经不对称加成反应合成了两个新型的Rh2(esp)2类似物——(Rs,Rs′,R,R′)-β-胺基羧酸酯(5)和(Rs,Rs′,R,R′,R,R′)-α,β-氮杂环丙烷羧酸酯(6, Scheme 1),产率分别为96%和65%,非对映选择性均大于20 ∶1。并对5进一步衍生获得了用于双核铑配位的双羧酸配体9。 5, 6和9均为新化合物,其结构经1H NMR和13C NMR表征。绝对构型根据文献报道中的单亚胺的反应立体控制得出[19]。
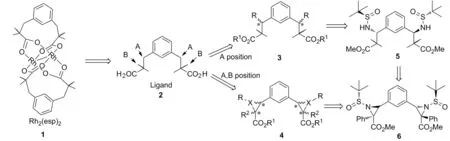
Chart 1
Scheme 1
1实验部分
1.1仪器与试剂
Varian Inova-400 MHz型核磁共振仪。
四乙氧基钛、(R)-叔丁基亚磺酰胺、对硝基苯磺酰氯、间苯二甲醛、苯乙酸甲酯,Alfa公司;无水THF,使用前用钠进行无水无氧处理并新蒸;二异丙胺使用前与氢化钙共热回流并新蒸;其余所用试剂均为分析纯。
1.2合成
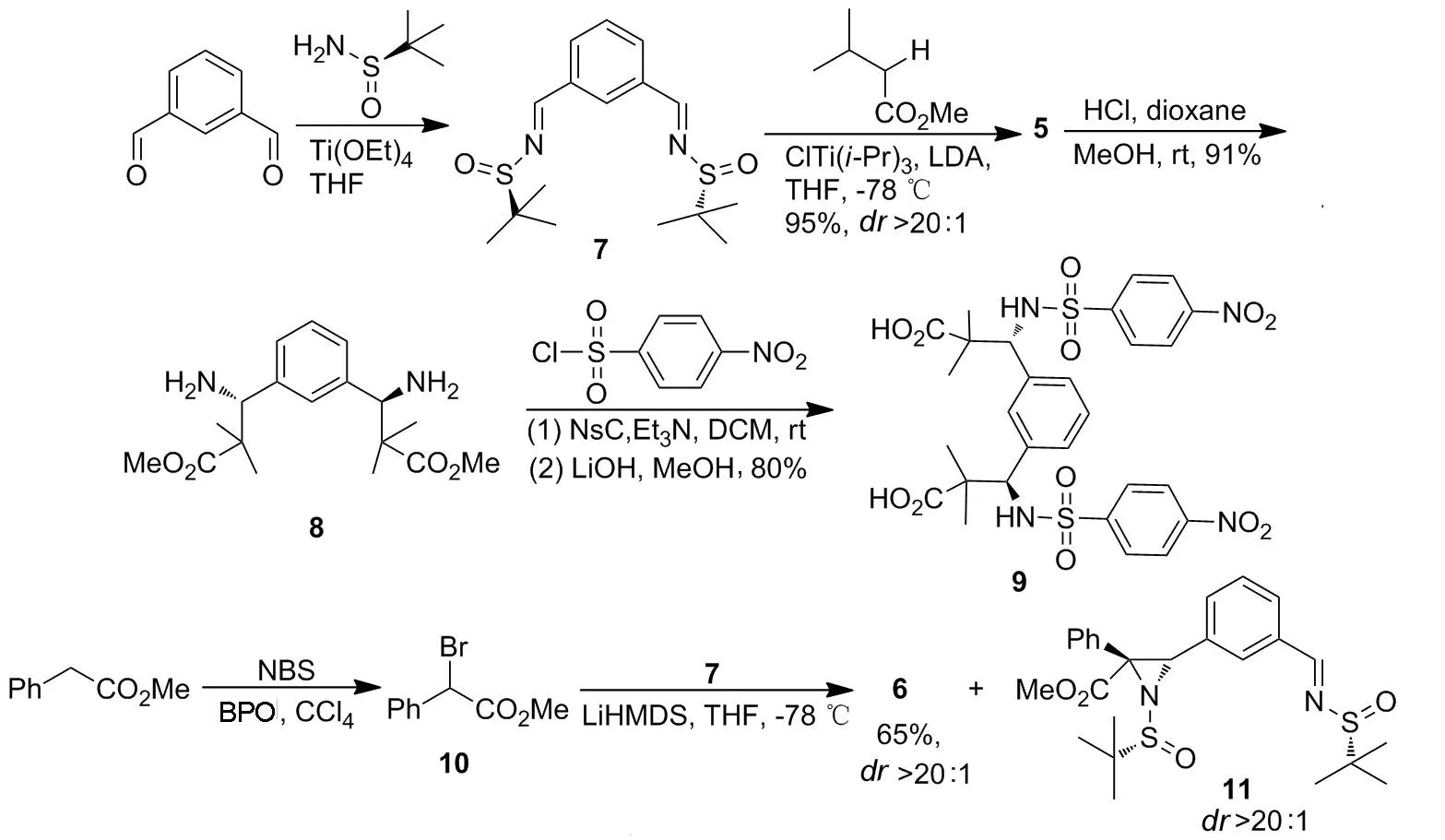
(1) 7的合成
在干燥的圆底烧瓶中依次加入Ti(OEt)411.63 g,间苯二甲醛1.37 g(10.2 mmol)和无水THF 80 mL,搅拌使其溶解;于室温反应30 min。加入 (R)-叔丁基亚磺酰胺2.73 g(22.5 mmol),回流反应5 h。冷却至室温,缓慢倒入冰水浴冷却过的饱和氯化钠溶液(80 mL)中,用硅藻土过滤,滤饼用乙酸乙酯(80 mL)洗涤,水相用乙酸乙酯(2×50 mL)萃取,合并滤液和萃取液,静置,用无水K2CO3干燥,浓缩后经硅胶柱层析 [洗脱剂:A=V(石油醚) ∶V(乙酸乙酯)=5 ∶1]纯化得黄色固体7 2.41 g,产率70%;1H NMRδ: 8.61(s, 2H), 8.28(s, 1H), 8.02~7.93(m, 2H), 7.60~7.56(m, 1H), 1.25(s, 18H);13C NMRδ: 161.9, 134.8, 132.8, 130.1, 129.7, 58.1, 22.7。
(2) 5的合成
氩气氛围下,在一干燥的Schlenk瓶中加入无水THF 360 mL,冰水浴冷却下用注射器注入二异丙胺5.2 mL;搅拌下缓慢加入2.5 mol·L-1正丁基锂溶液14.8 mL,反应30 min;于-78 ℃(浴温)滴加异丁酸甲酯4.2 mL(37.1 mmol),滴毕,反应1 h。缓慢滴加ClTi(Oi-Pr)319.09 g的无水THF (120 mL)溶液,滴毕,反应10 min。滴加8 2.08 g(6.1 mmol)的无水THF(10 mL)溶液,滴毕,反应1.5 h。缓慢加入饱和氯化铵溶液60 mL淬灭反应。升至室温,用硅藻土过滤,滤饼用氯仿100 mL洗涤;合并滤液与洗液,旋蒸除去THF,用氯仿(3×100 mL)萃取,合并有机相,用无水硫酸钠干燥,过滤,旋蒸除溶,剩余物用氯仿溶解,经硅胶柱层析[洗脱剂:B=V(二氯甲烷) ∶V(甲醇)=30 ∶1]纯化得淡黄色泥状物5 3.19 g,产率96%,dr>20 ∶1;1H NMRδ: 7.30~7.27(m, 2H), 7.19(s, 1H), 7.17~7.16(m, 1H), 4.58(d,J= 3.3 Hz, 2H), 4.47(d,J=3.3 Hz, 2H), 3.75(s, 6H), 1.19(s, 18H), 1.18(s, 6H), 1.17(s, 6H);13C NMRδ: 177.1, 138.0, 130.3, 129.0, 127.7, 64.8, 55.9, 52.6, 47.2, 24.4, 22.7, 20.8。
(3) 8的合成
在圆底烧瓶中加入5 5.43 g(10.0 mmol)和无水甲醇138 mL,搅拌使其溶解;缓慢加入4 mol·L-1盐酸二噁烷溶液25 mL,反应1.5 h。缓慢加入氨水80 mL淬灭反应;加水200 mL,用乙酸乙酯(2×250 mL)萃取,合并有机相,用饱和氯化钠溶液洗涤,无水硫酸钠干燥;旋蒸除溶后经硅胶柱层析(洗脱剂:B=30 ∶1)纯化得无色油状液体8 3.05 g,产率91%;1H NMRδ: 7.25~7.15(m, 4H), 4.22(s, 2H), 3.68(s, 6H), 1.71(br s, 2H), 1.12(s, 6H), 1.06(s, 6H);13C NMRδ: 177.8, 141.6, 128.3, 127.5, 127.3, 62.0, 52.0, 48.0, 23.9, 19.5。
(4) 9的合成
在一干燥反应茄形瓶中加入8 100 mg(0.30 mmol),对硝基苯磺酰氯166 mg(0.75 mmol)和无水二氯甲烷3.5 mL,搅拌使其溶解;滴加三乙胺109 μL,滴毕,于室温反应1 h。加入乙酸乙酯20 mL,依次用水(20 mL)和饱和氯化钠溶液(20 mL)洗涤,无水硫酸钠干燥;旋蒸除溶后用混合溶剂[V(甲醇) ∶V(水)=4 ∶1, 4 mL]溶解;加入无水氢氧化锂105 mg,回流反应18 h。冷却至室温,加水5 mL,用浓盐酸酸化,用乙酸乙酯(3×10 mL)萃取,合并有机相,用饱和氯化钠溶液(2×15 mL)洗涤,无水硫酸钠干燥,旋蒸除溶后经柱层析(洗脱剂:A=1 ∶1)纯化得白色固体9 163 mg,产率80%;1H NMRδ: 12.73(br s, 2H), 8.60(d,J=10.3 Hz, 2H), 7.99(d,J=8.7 Hz, 4H), 7.58(d,J=8.7 Hz, 4H), 6.79(s, 1H), 6.59(d,J=7.7 Hz, 2H), 6.40(t,J=7.7 Hz, 1H), 4.66(d,J=9.7 Hz, 2H), 1.02(s, 6H), 0.82(s, 6H);13C NMRδ:176.7, 148.7, 146.6, 135.9, 129.3, 127.9, 126.3, 126.0, 123.8, 62.9, 47.0, 21.9, 20.8。
(5) 10的合成
在圆底烧瓶中依次加入苯乙酸甲酯6.17 g(41.1 mmol),四氯化碳30 mL和过氧化苯甲酰(BPO)0.10 g,搅拌使其均匀;回流反应4 h。冷却至室温,过滤,滤饼用四氯化碳(30 mL)洗涤;合并滤液与洗液,旋蒸浓缩后经蒸馏(115 ℃/133 Pa)纯化得无色油状液体10 7.58 g,产率80%;1H NMRδ: 7.56~7.54(m, 2H), 7.40~7.34(m, 3H), 5.37(s, 1H), 3.79(s, 1H);13C NMRδ: 168.9, 135.8, 129.4, 128.9, 128.7, 53.5, 46.6。
(6) 6和11的合成
氩气氛围下,在干燥的Schlenk瓶中加入无水THF 65 mL和1.2 mol· L-1LiHMDS的THF溶液16.7 mL,搅拌下于-78 ℃加入7 4.58 g(20.0 mmol)的无水THF(5.0 mL)溶液,反应35 min。滴加7 0.68 g(2.0 mmol)的无水THF(20 mL)溶液,滴毕(30 min),反应1 h。缓慢加入饱和氯化铵溶液15 mL淬灭反应。升至室温,静置分层,水相用乙酸乙酯(2×100 mL)萃取,合并有机相和萃取液,用饱和氯化钠溶液(100 mL)洗涤,无水硫酸钠干燥,旋蒸除溶后经柱层析[梯度洗脱剂:A=3 ∶1~1 ∶1]纯化得6和11。
6: 白色固体,产率65%,dr>20 ∶1;1H NMRδ: 7.23~7.10(m, 6H), 6.95(d,J=6.6 Hz, 4H), 6.84(t,J=8.0 Hz, 1H), 6.69(d,J=6.4 Hz, 3H), 4.43(s, 2H), 3.75(s, 6H), 1.27(s, 18H);13C NMRδ: 169.3, 133.2, 132.7, 129.7, 127.8, 127.79, 127.76, 127.4, 126.9, 57.1, 54.3, 53.8, 52.3, 22.0。
11: 淡黄色固体,产率7%,dr>20 ∶1;1H NMRδ: 8.39(s, 1H), 7.62(d,J=7.7 Hz, 1H), 7.46(s, 1H), 7.23~7.14(m, 6H), 7.03(d,J=7.8 Hz, 1H), 4.65(s, 1H), 3.79(s, 3H), 1.31(s, 9H), 1.23(s, 9H);13C NMRδ: 169.4, 162.3, 134.4, 134.1, 132.5, 131.5, 129.8, 129.1, 128.8, 128.7, 128.1, 128.0, 58.0, 57.3, 54.5, 54.0, 52.4, 22.7, 22.0。
2结果与讨论
在5的合成中,ClTi(Oi-Pr)3影响反应过渡态的烯醇式构象以及烯醇锂盐对叔丁基亚磺酰亚胺的靠近方式[19],因而它的加入是获得高非对映选择性(dr>20 ∶1)反应产物的关键。原料最佳当量比为8∶(CH3)2CHCO2CH3∶LDA∶ClTi(i-Pr)3=1 ∶6 ∶6 ∶12。
在6的合成中,并不需要加入路易斯酸即可获得高立体选择性反应产物,但即便把10的用量提高到10 eq.以上,仍然有少量(7%)的11产生;继续延长反应时间也并不能使11完全转化为6。
此研究获得的手性双氨基酯存在亚磺酰基,硫上有一孤对电子,这对铑配位可能会造成不利的影响,诸如催化剂中毒等。为了避免这个问题的发生,我们对5进行了拓展衍生。用4 mol·L-1盐酸在二噁烷/甲醇体系中高效率地脱除叔丁基亚磺酰基而获得了双氨基酯8。接着经胺基官能化和水解的分步连续反应获得了双氨基酸化合物9。该结构中氨基上的氢具有一定的酸性,Du Bois J[20]的研究表明在手性内酰胺双核铑催化剂中,β-位磺酰胺基上的氢与酰胺的羰氧存在弱氢键作用,这个可能是使催化反应获得高立体选择性的关键。
手性叔丁基亚磺酰胺是合成含氮化合物的优良手性助剂,其与间苯二甲醛缩合所得的手性双亚胺可简便快捷地构建Rh2(esp)2配体的手性类似物,这一合成方法对于发展手性四羧酸双核铑催化剂有潜在的应用价值。
参考文献
[1]DeAngelis A, Boruta D T, Lubin J B,etal. The chiral in paddlewheel complexes[J].Chem Commun,2010,46:4541-4543.
[2]Doyle M P, McKervey M A, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds:From Cyclopropanes to Ylides[M].New York:John Wiley & Sons,Inc.,1998:1-46.
[3]Evans P A. Modern Rhodium-catalyzed Organic Reactions[M].Weinheim:Wiley-VCH,2005:341-353.
[4]Davies H M L, Manning J R. Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion[J].Nature,2008,451:417-424.
[5]Crabtree R H. Introduction toselective functionalization of C-H bonds[J].Chem Rev,2010,110:575-575.
[6]Davies H M L, Du Bois J, Yu J Q. C-H functionalization in organic synthesis[J].Chem Soc Rev,2011,40:1855-1856.
[7]Espino C G, Fiori K W, Du Bois J,etal. Expanding thescope of C-H amination through catalyst design[J].J Am Chem Soc,2004,126:15378-15379.
[8]Bess E N, DeLuca R J, Tindall D J,etal. Analyzing site selectivity in Rh2(esp)2-catalyzed intermolecular C-H amination reactions[J].J Am Chem Soc,2014,136:5783-5789.
[9]Du Bois J. Rhodium-catalyzed C-H amination.An enabling method for chemical synthesis[J].Org Process Res Dev,2011,15:758-762.
[10]Zalatan D N, Du Bois J. Understanding the differential performance of Rh2(esp)2as a catalyst for C-H amination[J].J Am Chem Soc,2009,131:7558-7559.
[11]Fiori K W, Du Bois J. Catalytic intermolecular amination of C-H bonds:Method development and mechanistic insights[J].J Am Chem Soc,2007,129:562-568.
[12]Reddy R P, Davies H M L. Dirhodium tetracarboxylates derived from adamantylglycine as chiral catalysts for enantioselective C-H aminations[J].Org Lett,2006,8:5013-5016.
[13]Liang C, Collet F, Dauban P,etal. Toward a synthetically useful stereoselective C-H amination of hydrocarbons[J].J Am Chem Soc 2008,130:343-350.
[14]Bickley J, Bonar-Law R, Steiner A,etal. Dirhodium (Ⅱ) carboxylate complexes as building blocks.Cis-chelating dicarboxylic acids designed to bridge the dinuclear core[J].New J Chem,2004,28:425-433.
[15]Robak M T, Herbage M A, Ellman J A. Synthesis and applications of tert-butanesulfinamide[J].Chem Rev,2010,110:3600-3740.
[16]Bo L, Lu C D. Asymmetric synthesis ofcis-2-aminocyclopropanols by intramolecular mannich addition of silyloxy benzyl carbanions[J].J Org Chem,2011,76:4205-4209.
[17]Ming Y, Lu C D. Three-component reactions of sulfonylimidates,silyl glyoxylates andN-tert-butanesulfinyl aldimines:An efficient,diastereoselective,and enantioselective synthesis of cyclicN-sulfonylamidines[J].Org Lett,2011,13:2782-2785.
[18]JiangJ L, Ming Y, Lu C D. Efficient synthesis ofα-quaternaryα-hydroxy-β-amino esters via silyl glyoxylate-mediated three-component reactions[J].Org Lett,2013,16:318-321.
[19]Tang T P, Ellman J A. Asymmetric synthesis ofα-amino acid derivatives incorporating a broad range of substitution patterns by enolate additions to tert-butyl sulfinyl Imines[J].J Org Chem,2002,67:7819-7832.
[20]Zalatan D N, Du Bois J. A chiral rhodium carboxamidate catalyst for enantioselective C-H amination[J].J Am Chem Soc,2008,130:9220-9221.
Synthesis of Novel Enantioenriched Analogues of Rh2(esp)2Ligand
HUANG Ze-ao1,2,LU Chong-dao1*
(1. Xinjiang Technical Institute of Physics & Chemistry, Chinese Academy of Sciences, Urumqi 830011, China;2. University of Chinese Academy of Science, Beijing 100049, China)
Abstract:A synthetic approach toward the enantioenriched analogues of α,α,α′,α′-tetramethyl-1,3-benzenedipropionic acid which was the ligand of Rh2(esp)2 catalyst was reported. In this process, the chiral (R)-tert-butanesulnyl diimine derived from 1,3-benzenedialdehyde was used in the nucleophilic additions of lithium ester enolates to give two novel chiral analogues of the “esp” esters that are (Rs,Rs′,R,R′)-β-amino esters and (Rs,Rs′,R,R′,R,R′)-α,β- aziridinyl esters in yields of 96% and 65% with excellent diastereoselectivity(dr>20 ∶1). The structures were characterized by1H NMR and (13)C NMR.
Keywords:Rh2(esp)2; dirhodium catalyst; tert-butanesulnyl diimine; (Rs,Rs′,R,R′)-β-amino ester; (Rs,Rs′,R,R′,R,R′)-α,β- aziridinyl ester; synthesis; chiral modification; diastereoselectivity
中图分类号:O621.3; O622.5
文献标志码:A
DOI:10.15952/j.cnki.cjsc.1005-1511.2016.03.15115
作者简介:黄泽傲(1987-),男,壮族,广西武宣人,博士研究生,主要从事有机金属催化剂的设计、合成及其催化C-H官能化的研究。 E-mail: huangzeao@yahoo.com通信联系人: 卢崇道,博士,研究员,博士生导师, E-mail: clu@ms.xjb.ac.cn
基金项目:国家自然科学基金资助项目(21172251)
收稿日期:2015-04-16;
修订日期:2016-01-28
