Substance P and its tachykinin NK1 receptor: a novel neuroprotective target for Parkinson’s disease
2015-12-15EmmaThornton,RobertVink
Substance P and its tachykinin NK1 receptor: a novel neuroprotective target for Parkinson’s disease
Parkinson’s disease (PD) is the most common motor neurodegenerative disorder aff ecting approximately 4 million people worldwide. Although PD presents primarily with motor dysfunction, non-motor symptoms including cognitive decline, mood disorders, reduced olfaction and constipation are also often present, with some of these non-motor symptoms even presenting prior to the onset of motor symptoms. It is well known that PD is largely caused by the gradual degeneration of dopaminergic neurons within the substantia nigra pars compacta (SNc), along with the presence of protein aggregates called Lewy bodies, which consist primarily of α-synuclein and are found in the cytoplasm of surviving neurons. This ongoing cell loss and Lewy body pathology is not confi ned to the SNc, but is also seen in other brain regions implicated in PD pathogenesis such as the locus ceruleus. Nonetheless, the cause of cell death in PD still remains somewhat elusive. Numerous secondary cell injury cascades have been linked to this progressive dopaminergic cell death, including infl ammation, blood brain barrier (BBB) dysfunction, oxidative stress, glutamate excitotoxicity and mitochondrial dysfunction. Despite this, current PD treatment does not protect dopaminergic neurons but rather provides symptomatic benefi t by replacing lost striatal dopamine (DA) either directly or by preventing its breakdown. The “gold-standard”treatment for PD is L-3,4-dihydroxyphenylalanine (L-DOPA), the precursor to DA, combined with a peripheral dopa decarboxylase inhibitor such as benseraside. Since its inception as a PD therapeutic over 50 years ago, L-DOPA has primarily been administered in a pulsatile manner. This type of L-DOPA delivery results in maladaptive changes to brain signaling pathways and expression of L-DOPA induced dyskinesia (LID), or involuntary abnormal movements. Unfortunately, all patients will develop LID at some stage throughout their treatment, which they often fi nd much more debilitating than the motor symptoms they presented with at diagnosis. Current research is thus focused on developing novel neuroprotective therapies that aim to treat the underlying cause of the disease, as well discovering possible adjunct therapies to L-DOPA that inhibit the expression of LID without interfering with L-DOPA motor improvement. Our recent studies have identifi ed the neuropeptide substance P (SP) and its tachykinin NK1 receptor (NK1-R) as novel neuroprotective targets for both ongoing dopaminergic degeneration and mild to moderate LID. Thus treatment with an NK1-R antagonist may provide a new PD therapeutic that shows particular effi cacy in the early stages of PD and LID (Thornton and Vink, 2012; Thornton et al., 2014).
Substance P and NK1-R are widely expressed throughout the peripheral and central nervous systems in primary sensory neurons or C-fi bers, as well as on endothelial cells and infl ammatory cells. They are expressed in particularly high levels within the SNc where SP-containing striatal projection neurons synapse with dopaminergic neurons to potentiate the release of striatal DA, normal levels of which are required for proper motor function. It is the death of dopaminergic neurons and the subsequent loss of dopaminergic drive in this system that results in the loss of nigral SP expression seen post-mortem, and in experimental models of PD where near total degeneration of dopaminergic neurons occurs. This contrasts to early PD where remaining dopamine neurons can maintain striatal DA levels. At this time, nigral SP expression is unaff ected or even increased. Indeed in our studies, SP expression was elevated following treatment with 6-hydroxydopamine (6-OHDA) in vitro, with a significant correlation between SP levels and cell death (Thornton et al., 2010). Similarly, SP expression was increased in rodents following intratstriatal injections of 6-OHDA as early as day 3 in the striatum and substantia nigra, which was prior to the onset of dopaminergic degeneration that began around day 7 post-lesion (Thornton and Vink, 2012). Moreover when intrastriatal 6-OHDA injections were combined with endogenous SP treatment, signifi cantly greater dopaminergic degeneration and further loss of motor function was seen compared to vehicle treated animals. In contrast, combination treatment of an NK1-R antagonist with 6-OHDA prevented the increase in SP seen at day 21 post-6-OHDA, and protected dopaminergic neurons and improved motor function. These results suggest that SP may be important in the early stages of PD pathogenesis.
This early increase of SP expression in PD may not only be due to a greater release from striatal projection neurons to maintain striatal DA levels, but may also be due to dopaminergic degeneration initiating neurogenic infl ammation, a neurally elicited local infl ammatory response. Neurogenic infl ammation occurs in the setting of injury or infl ammation, both of which are present in PD, and results in the release of SP and calcitonin gene-related peptide (CGRP) from C-fi bers surrounding blood vessels. Calcitonin gene-related peptide, a potent vasodilator, facilitates recruitment of infl ammatory mediators, whereas SP binds to NK1-R on endothelial cells of cerebral microvasculature to increase BBB permeability. Normally, the structure of the BBB confers low paracellular permeability because of specialized endothelial cells with low pinocytotic activity, and expression of interendothelial adheren and tight junctional proteins such as cadherins, claudins, occludin and junctional adhesion molecules. This structural arrangement ensures that the passage of substances through the cerebral microvasculature is highly controlled, which is vital to maintain neuronal homeostasis (Hawkins and Davis, 2005). Consequently, dysfunction of the BBB stimulates an unfavorable neuronal environment due to the presence of normally excluded proteins, ions, toxins, metals and cells.
Increased permeability of the BBB has been observed in clinical and animal models of PD with decreased expression of the effl ux transporter P-glycoprotein and increased extravasation of the serum protein albumin being reported (Kortekaas et al., 2005; Pisani et al., 2012). Whether these changes are due to increased protein transport or loss of structural proteins like claudin-5 or occludin remains to be elucidated. Of interest for PD pathogenesis is that the barrier is particularly weak surrounding the cerebral microvasculature of the substantia nigra, with dopaminergic neurons seeming to be more vulnerable to experimentally induced barrier dysfunction than other neuronal populations (Rite et al., 2007). This may be due to the increased basal level of oxidative stress in the SNc from the formation of reactive oxygen species (ROS) during normal DA metabolism, as oxidative stress can in itself induce BBB dysfunction and has been strongly linked to dopaminergic cell death. It is this abnormally high level of oxidative stress in the substantia nigra that has been attributed to the increased albumin to cerebrospinal
fl uid ratio seen in the healthy elderly compared to the young and healthy population. Therefore in PD, where age is the greatest risk factor, the BBB may, in part, already to be functioning incorrectly. This means that only minor changes to the cerebral environment are required to induce further impairment of the barrier and subsequent dopaminergic cell loss.
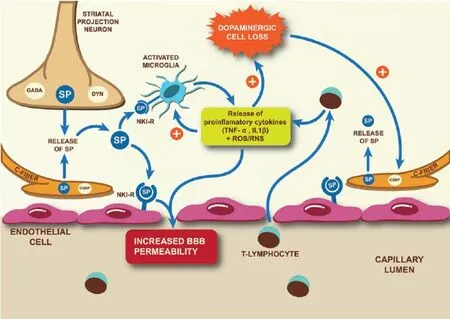
Figure 1 Mechanisms by which SP-induced neurogenic infl ammation is implicated in the pathogenesis of Parkinson’s disease.
BBB dysfunction is also thought to contribute to LID by facilitating the uncontrolled entry of L-DOPA from the blood into the brain parenchyma resulting in fl uctuating endogenous DA levels. Interestingly, administration of L-DOPA re-instates the dopaminergic drive in the SP-DA positive feedback regulation, increasing nigral SP expression back to normal levels. In doing so, endogenous SP is once again available to induce barrier breakdown. Indeed, L-DOPA treatment has been observed to increase barrier permeability to albumin (Westin et al., 2006). Apart from a direct ability to increase BBB permeability through neurogenic infl ammation, SP is also involved in angiogenesis. Formation of new blood vessels with immature BBB properties has been observed in experimental LID (Westin et al., 2006). Due to the lack of an eff ective barrier on these vessels, L-DOPA delivery to the brain is unregulated causing fl uctuating endogenous DA levels and LID. Confi rmation that SP may be involved in the onset of LID was fi rst reported in our studies where NK-1R antagonist treatment prevented the onset of mild to moderate LID (Thornton et al., 2014). Although the exact mechanism of this protection was not studied, the reduced FosB levels suggested regulation of the D1-mediated basal ganglia signaling pathway and prevention of the maladaptive changes. This could have been partially due to the NK1-R antagonist stabilizing the barrier, resulting in regulated delivery of DA to striatal projections neurons (Thornton et al., 2014). This role for SP in the onset of LID was recently confi rmed by Yang et al. (2015), who showed treatment with an NK1-R antagonist reduced LID in 6-OHDA lesioned rodents by preventing DA effl ux and the subsequent increase in secondary messenger systems, Arc and Penk (Yang et al., 2015). Importantly, both studies confi rmed that administration of the NK1-R antagonist did not interfere with L-DOPA induced improvement in motor function. This augurs well for the use of NK1-R antagonists as a novel adjunct therapy to L-DOPA in the treatment of PD.
Along with its eff ects on endothelial cells and BBB permeability, SP can also directly interact with infl ammatory cells by binding to NK1-R expressed on microglia, astrocytes and leukocytes resulting in mast cell degranulation and cytokine production.
Interestingly, histamine, which is released during mast cell degranulation, has been associated with BBB breakdown and the degeneration of dopaminergic neurons (Liu et al., 2007). Infl ammatory processes in PD are unfortunately self-perpetuating with dying neurons activating the resident immune cells, including microglia. In turn, microglia produce high levels of ROS including superoxide, reactive nitrogen species (RNS) and pro-infl ammatory cytokines resulting in further degeneration of dopaminergic neurons. This results in a vicious cycle of ongoing cell death and chronic infl ammation. Indeed chronic activation of microglia has been visualized using positron emission tomography in the midbrain of PD patients and is a consistent feature in animal models of PD. Infl ammatory processes are exacerbated in PD by the infiltration of peripheral immune cells such as T-lymphocytes, which are able to enter through the dysfunctional barrier and release pro-infl ammatory cytokines, thereby further activating resident immune cells. Indeed, CD4+and CD8+T-cells have been observed within the SN in both the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of PD and at postmortem, with CD4+T cells linked to MPTP-induced dopaminergic neurodegeneration (Brochard et al., 2009). The release of pro-infl ammatory factors from either resident or peripheral immune cells may also directly increase BBB permeability by changing the expression of tight junction proteins (as reviewed by Hawkins and Davis, 2005) thereby allowing further infi ltration of unwanted substances that could infl uence neurodegeneration. These results suggest that BBB dysfunction and infl ammation work in concert to exacerbate ongoing dopaminergic cell death.
Our own studies examining the relationship between BBB dysfunction and inflammatory processes in the 6-OHDA intrastriatal model of PD have confi rmed their association with dopaminergic cell death (Thornton and Vink, 2012). In these studies, progressive dopaminergic degeneration was associated with both albumin extravasation and increased numbers of resident infl ammatory cells, with both barrier disruption and activated microglia peaking at day 14 post-lesion coincident with signifi cant dopaminergic degeneration. Treatment with an NK1-R antagonist prevented the albumin extravasation, and was accompanied by a reduction in infl ammation. Although peripheral immune cell infi ltration was not directly measured in this study, ED-1, a marker of phagocytotic cells including activated microglia and peripheral blood-borne macrophages, was assessed. NK1-R antagonist treatment did cause a reduction in the number of nigral ED-1 positive cells, which could represent a reduction in the direct activation of microglia and/or prevention of peripheral blood-borne macrophage recruitment due to restoration of BBB integrity.
In summary, SP-induced neurogenic infl ammation may play an important role in the pathogenesis of PD, including LID, by increasing BBB permeability and subsequent peripheral immune cell infi ltration, as well as activation of resident immune cells (Figure 1). These injury cascades not only directly contribute to the ongoing neurodegeneration of dopaminergic neurons in PD but can also exacerbate other secondary injury mechanisms such as oxidative stress and mitochondrial dysfunction. Treatment with an NK1-R antagonist attenuated the progression of PD and signifi cantly reduced the onset of LID, presumably by inhibiting multifactorial injury mechanisms involving SP and NK1-R. Blocking the eff ects of SP with a NK1-R antagonist therefore presents a potentially promising new avenue for neuroprotection in PD.
This work has been presented at the International Congress of Parkinson’s Disease and Movement Disorders (2009 and 2010), and in part has been supported by the Neurosurgical Research Foundation, South Australia, Australia. The authors would like to thank Tavik Morgenstern for assistance with the Figure 1 graphics. Emma Thornton, Robert Vink*
School of Medical Sciences, University of Adelaide, Adelaide, Australia; Division of Health Sciences, University of South Australia, Adelaide, Australia
*Correspondence to: Robert Vink, Ph.D.,
Robert.Vink@unisa.edu.au.
Accepted: 2015-08-10
orcid: 0000-0002-4885-0667 (Robert Vink)
Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, Duyckaerts C, Flavell RA, Hirsch EC, Hunot S (2009) Infi ltration of CD4+lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest 119:182-192.
Hawkins BT, Davis TP (2005) The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev 57:173-185.
Kortekaas R, Leenders KL, van Oostrom JC, Vaalburg W, Bart J, Willemsen AT, Hendrikse NH (2005) Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol 57:176-179.
Liu CQ, Chen Z, Liu FX, Hu DN, Luo JH (2007) Involvement of brain endogenous histamine in the degeneration of dopaminergic neurons in 6-hydroxydopamine-lesioned rats. Neuropharmacology 53:832-841.
Pisani V, Stefani A, Pierantozzi M, Natoli S, Stanzione P, Franciotta D, Pisani A (2012) Increased blood-cerebrospinal fl uid transfer of albumin in advanced Parkinson’s disease. J Neuroinfl ammation 9:188.
Rite I, Machado A, Cano J, Venero JL (2007) Blood-brain barrier disruption induces in vivo degeneration of nigral dopaminergic neurons. J Neurochem 101:1567-1582.
Thornton E, Vink R (2012) Treatment with a substance P receptor antagonist is neuroprotective in the intrastriatal 6-hydroxydopamine model of early Parkinson’s disease. PLoS One 7:e34138.
Thornton E, Tran TT, Vink R (2010) A substance P mediated pathway contributes to 6-hydroxydopamine induced cell death. Neurosci Lett 481:64-67.
Thornton E, Hassall MM, Corrigan F, Vink R (2014) The NK1 receptor antagonist N-acetyl-L-tryptophan reduces dyskinesia in a hemi-parkinsonian rodent model. Parkinsonism Relat Disord 20:508-513.
Westin JE, Lindgren HS, Gardi J, Nyengaard JR, Brundin P, Mohapel P, Cenci MA (2006) Endothelial proliferation and increased blood-brain barrier permeability in the basal ganglia in a rat model of 3,4-dihydroxyphenyl-L-alanine-induced dyskinesia. J Neurosci 26:9448-9461.
Yang X, Zhao H, Shi H, Wang X, Zhang S, Zhang Z, Zu J, Zhang W, Shen X, Cui G, Hua F (2015) Intranigral administration of substance P receptor antagonist attenuated levodopa-induced dyskinesia in a rat model of Parkinson’s disease. Exp Neurol 271:168-174.
10.4103/1673-5374.165505 http://www.nrronline.org/
Thornton E, Vink R (2015) Substance P and its tachykinin NK1 receptor: a novel neuroprotective target for Parkinson’s disease. Neural Regen Res 10(9):1403-1405.
杂志排行

中国神经再生研究(英文版)的其它文章
- PTEN inhibition and axon regeneration and neural repair
- Neural correlates of the Heidelberg Music Therapy: indicators for the regeneration of auditory cortex in tinnitus patients?
- The choline pathway as a strategy to promote central nervous system (CNS) remyelination
- Enhancing endogenous stem cells in the newborn via delayed umbilical cord clamping
- Elastic modulus aff ects the growth and diff erentiation of neural stem cells
- Non-steroidal anti-infl ammatory drugs (NSAIDs) and neuroprotection in the elderly: a view from the mitochondria