Non-steroidal anti-infl ammatory drugs (NSAIDs) and neuroprotection in the elderly: a view from the mitochondria
2015-12-15MariaCalvo-Rodríguez,LucíaNú?ez,CarlosVillalobos
Non-steroidal anti-infl ammatory drugs (NSAIDs) and neuroprotection in the elderly: a view from the mitochondria
The most important risk factor for stroke and neurodegeneration is aging. In fact, survival after stroke diminishes largely with aging. In fact, recovery after brain artery occlusion is dramatically worsened by aging, even normal aging is associated with neuron damage and cognitive decline. Mechanisms involved in aging-related, cognitive decline and susceptibility to neuron damage in stroke and neurodegeneration are largely unknown. One of the most important mechanisms contributing to neural dysfunction and death is excitotoxicity. This process is based on the fact that the excessive glutamate receptor stimulation may lead to neuronal damage. This overstimulation may be due to increased concentration of glutamate, or the prolonged activation of receptors.
Protecting the aging brain against damage remains a big challenge for neurologists and neuroscientists. Interestingly, a large number of basic and clinical studies have provided strong evidence indicating that the prolonged use of non-steroidal anti-inflammatory drugs (NSAIDs) may reduce the incidence of Alzheimer’s disease (AD) (Wang et al., 2015), the most common form of dementia. NSAIDs also decreased glutamate excitotoxicity both in vitro, in rat primary neuronal cultures and hippocampal slices (Grilli et al., 1996), and in vivo, protecting rats against rotenone-induced parkinsonism (Madathil et al., 2013). Recent evidence suggests also that NSAIDs may even protect against the cognitive decline associated to healthy aging in humans (Kern et al., 2012).
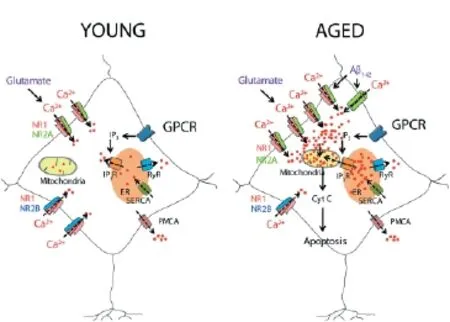
Figure 1 A model of neurotoxicity induced by excessive Ca2+entry induced by glutamate or by Aβ1–42oligomers in aged neurons.
NSAIDs present antipyretic, anti-inflammatory and analgesic eff ects. Therefore, they are mainly used to relief pain, fever and inflammation. Their best characterized action is inhibition of cyclooxygenase (COX), and thus the synthesis of prostaglandins, which participate in the inflammatory response. NSAIDs can be non-selective COX inhibitors such as aspirin, ibuprofen, indomethacin or sulindac; or selective COX-2 inhibitors, such as rofecoxib and celecoxif. The action mechanism of neuroprotection by NSAIDs is unknown, but reports suggest that it is not related to the classic anti-infl ammatory activity of these drugs. It is widely accepted that neuronal excitotoxicity induced by glutamate is mainly caused by one kind of ionotropic glutamate receptor, the N-methyl-D-aspartate receptor (NMDAR), probably because of its high permeability to Ca2+. The combination of diff erent subunits constitutes NMDARs: The NR1 subunit, is ubiquitous and essential whereas the NR2 subunit, is a regulatory subunit (NR2A - NR2D). There is also a third subunit named NR3 (NR3A - NR3B). A functional NMDA receptor requires the binding of two NR1 subunits with two other NR2 subunits or with the combination of a NR2 and NR3 subunits. In normal synaptic transmission, the NMDAR, blocked by the Mg2+located in the channel, is activated for short periods of time.
However, in pathological conditions, like the prolonged depolarization that takes place in ischemic events, during which Mg2+is fully removed from its binding site at the NMDARs, an overly activation of the receptor causes excessive Ca2+entry through the channel. This Ca2+entry, together with the Ca2+released from the intracellular stores, increases the cytosolic free Ca2+concentration to levels that exceed the capacity of the intracellular Ca2+clearing mechanisms and pumps leading to mitochondrial Ca2+overload. This may cause impaired metabolism and certain processes that trigger cell death such as the one in the neurodegenerative disorders (Pivovarova et al., 2004).
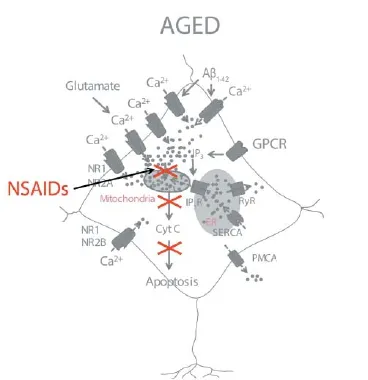
Figure 2 A model of NSAID neuroprotection based on the inhibition of mitochondrial Ca2+overload.
We have reported that oligomers, but not fi brils, of the amyloid β peptide 1–42 (Aβ1–42), the most likely toxin in AD, induce also a sustained entry of Ca2+followed by mitochondrial Ca2+overload leading to cell death in cultures of rat cerebellar granule cells (Sanz-Blasco et al., 2008). The pathway for Ca2+entry remains unknown, but several reports suggest it could be mediated, at least partially, by NMDA receptors. Interestingly, we showed that a series of NSAIDs, including salicylate (the major metabolite of aspirin), ibuprofen, sulindac sulfi de, indomethacin and the structural analogue lacking anti-inflammatory activity R-flurbiprofen, are able to depolarize
partially mitochondria preventing mitochondrial Ca2+overload without aff ecting Ca2+infl ux induced by oligomers of Aβ1–42. All these eff ects were achieved at fairly low concentrations of NSAIDs, in the μM range, far from those required for preventing infl ammation or reducing the Aβ burden. Mitochondrial depolarization could be easily explained by the chemical structure of carboxylic NSAIDs, resembling mild mitochondrial uncouplers. These class of compounds are able to decrease partially mitochondrial potential, the huge driving force for mitochondrial Ca2+uptake, in a similar manner to low concentrations of established mitochondrial uncouplers as carbonyl cyanide-p-trifl uoromethoxyphenylhydrazone (FCCP).
Understanding the mechanisms of aging-related susceptibility to neuron damage and neuroprotection by NSAIDs is critical since they may provide feasible ways of preventing brain damage in the elderly. However, this study has been hampered by the lack of suitable models of brain aging. Recently, several authors have provided evidence indicating that long-term cultures of hippocampal neurons show many of the typical hallmarks of aged neurons including accumulation of reactive oxygen species (ROS), lipofuscin granules, heterochromatic foci, activation of the Jun N-terminal protein kinase (pJNK) and p53/p21 pathways, gradual loss of cholesterol, and changes in Ca2+channel density and NMDA receptor expression (Sodero et al., 2011). In addition, studies show the increased vulnerability of hippocampal neurons with age in culture (Brewer et al., 2007). We have recently used long-term cultures of neonatal rat hippocampal neurons to investigate age-related susceptibility to excitotoxicity and neuroprotection by NSAIDS. We found that NMDA promoted cell death only in aged neurons cultured for several weeks in vitro but not in young cultures (Calvo et al., 2015), despite both expressed NMDA receptors and showed NMDA-induced rises in cytosolic [Ca2+]. However, the increases in cytosolic [Ca2+] induced by NMDA were much larger in older cultures than in younger neurons. These changes correlated with changes in the composition and density of NMDA receptor subunits consistently with those observed in vivo (Cui et al., 2013). Most importantly, NMDA induced mitochondrial Ca2+uptake only in aged neurons. Thus, mitochondria from young neurons are seemingly not sensitive to the changes in cytosolic [Ca2+].
Several mechanisms may contribute to diff erential behavior of mitochondria from young and old neurons, but the most likely one is the increased rises in cytosolic [Ca2+] and enhanced resting cytosolic [Ca2+] observed in older neurons (Calvo et al., 2015). Whereas the mechanism for enhanced resting cytosolic [Ca2+] remains unknown, the basis for increased responses to NMDA can be explained by the changes in NMDA receptor density and composition (Calvo et al., 2015). In summary, mitochondrial Ca2+overload is critical for enhanced susceptibility to cell death in aged neurons (Figure 1). This view is supported by the fact that NMDA promotes permeability transition and release of cytochrome c in older neurons and these eff ects are prevented by mitochondrial depolarization with FCCP. The role of mitochondria is further supported by the fact that several NSAIDs, including salicylate, indomethacin, sulindac sulphide and the structural analogue R-fl urbiprofen depolarize mitochondria and prevent also mitochondrial Ca2+uptake and neuron cell death without aff ecting the rise in cytosolic [Ca2+] induced by NMDA. Therefore, these results strongly suggest that enhanced mitochondrial Ca2+uptake contributes largely to aging-related susceptibility to neuron cell damage and cognitive decline. Most importantly, they also support the view that these processes could be prevented to some extent by the use of low concentrations of NSAIDs (Figure 2). Of course, clinical research is required to fully support this view.
Interestingly, some NSAIDs have been already proposed or even tested to prevent neuron cell death in brain pathologies. For example, R-fl urbiprofen (FlurizanTM) was tested for preventing AD in a large-scale clinical trial that failed in its phase three. Notably, this compound was used at large concentrations aimed at reducing the Aβ burden. The reasons for the failure of R-fl urbiprofen in this trial are not clear at present but it has been proposed that damage in AD patients might be too severe to be reversed by even the best drugs. In support of this view, it has been shown recently that R-fl urbiprofen, a drug that lacks anti-infl ammatory activity, prevents and attenuates primary progressive experimental multiple sclerosis in mice, even if the treatment commenced on or after the fi rst signs of the disease (Schmitz et al., 2014). Further research is required to test the use of selected NSAIDs and structural analogues without anti-infl ammatory activity in neuron damage associated to aging. This work was supported by grants VA145U13, BIO/VA33/13, BIO103/VA45/11 from Junta de Castilla y León, Spain and BFU2012-37146 from Ministerio de Economía y Competitividad, Spain. MCR was supported by a pre-doctoral fellowship from Junta de Castilla y León, Spain and The European Social Fund.
María Calvo-Rodríguez, Lucía Núñez, Carlos Villalobos*
Instituto de Biología y Genética Molecular (IBGM), Consejo Superior de Investigaciones Científi cas (CSIC) and Universidad de Valladolid, c/ Sanz y Fores 3, 47003 Valladolid, Spain
*Correspondence to: Carlos Villalobos, Ph.D., carlosv@ibgm.uva.es.
Accepted: 2015-06-10
Brewer LD, Thibault O, Staton J, Thibault V, Rogers JT, Garcia-Ramos G, Kraner S, Landfield PW, Porter NM (2007) Increased vulnerability of hippocampal neurons with age in culture: temporal association with increases in NMDA receptor current, NR2A subunit expression and recruitment of L-type calcium channels. Brain Res 1151:20-31.
Calvo M, Sanz-Blasco S, Caballero E, Villalobos C, Nunez L (2015) Susceptibility to excitotoxicity in aged hippocampal cultures and neuroprotection by non-steroidal anti-infl ammatory drugs: role of mitochondrial calcium. J Neurochem 132:403-417.
Cui Z, Feng R, Jacobs S, Duan Y, Wang H, Cao X, Tsien JZ (2013) Increased NR2A:NR2B ratio compresses long-term depression range and constrains long-term memory. Sci Rep 3:1036.
Grilli M, Pizzi M, Memo M, Spano P (1996) Neuroprotection by aspirin and sodium salicylate through blockade of NF-kappaB activation. Science 274:1383-1385.
Kern S, Skoog I, Ostling S, Kern J, Borjesson-Hanson A (2012) Does lowdose acetylsalicylic acid prevent cognitive decline in women with high cardiovascular risk? A 5-year follow-up of a non-demented population-based cohort of Swedish elderly women. BMJ Open 2.
Madathil SK, Karuppagounder SS, Mohanakumar KP (2013) Sodium salicylate protects against rotenone-induced parkinsonism in rats. Synapse 67:502-514.
Pivovarova NB, Nguyen HV, Winters CA, Brantner CA, Smith CL, Andrews SB (2004) Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. J Neurosci 24:5611-5622.
Sanz-Blasco S, Valero RA, Rodriguez-Crespo I, Villalobos C, Nunez L (2008) Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS One 3:e2718.
Schmitz K, de Bruin N, Bishay P, Mannich J, Haussler A, Altmann C, Ferreiros N, Lotsch J, Ultsch A, Parnham MJ, Geisslinger G, Tegeder I (2014) R-fl urbiprofen attenuates experimental autoimmune encephalomyelitis in mice. EMBO Mol Med 6:1398-1422.
Sodero AO, Weissmann C, Ledesma MD, Dotti CG (2011) Cellular stress from excitatory neurotransmission contributes to cholesterol loss in hippocampal neurons aging in vitro. Neurobiol Aging 32:1043-1053.
Wang J, Tan L, Wang HF, Tan CC, Meng XF, Wang C, Tang SW, Yu JT (2015) Anti-infl ammatory drugs and risk of Alzheimer’s disease: an updated systematic review and meta-analysis. J Alzheimers Dis 44:385-96.
10.4103/1673-5374.165219 http://www.nrronline.org/
Calvo-Rodríguez M, Núñez L, Villalobos C (2015) Non-steroidal anti-infl ammatory drugs (NSAIDs) and neuroprotection in the elderly: a view
from the mitochondria. Neural Regen Res 10(9):1371-1372.
杂志排行

中国神经再生研究(英文版)的其它文章
- PTEN inhibition and axon regeneration and neural repair
- Neurochemical plasticity of Müller cells after retinal injury: overexpression of GAT-3 may potentiate excitotoxicity
- The choline pathway as a strategy to promote central nervous system (CNS) remyelination
- Enhancing endogenous stem cells in the newborn via delayed umbilical cord clamping
- Elastic modulus aff ects the growth and diff erentiation of neural stem cells
- Neural correlates of the Heidelberg Music Therapy: indicators for the regeneration of auditory cortex in tinnitus patients?