C6H6/C2H2…(HX)2(X=F,Cl,Br,I)分子间相互作用研究
2015-11-23赵培培李少波席小倩
阳 杰 叶 青 肖 扬 赵培培 张 霞 李少波 叶 朗 席小倩
(合肥学院,安徽 合肥 230022)
C6H6/C2H2…(HX)2(X=F,Cl,Br,I)分子间相互作用研究
阳 杰 叶 青 肖 扬 赵培培 张 霞 李少波 叶 朗 席小倩
(合肥学院,安徽 合肥 230022)
采用从头算MP2方法,对乙炔分子中的π键与(HX)2(X=F,Cl,Br,I)形成的典型X-H···π键复合物进行了系统的研究,并对构建的复合物结构、相互作用能和BSSE进行了优化计算,对计算结果在结构、分子间相互作用能等参数进行分析,最后对C6H6中的π键与(HCl)2形成的典型X-H…π键复合物进行计算研究,并比较炔烃和芳香烃中的两种π键与卤化氢形成的复合物在氢键性质差异。研究结果表明,C2H2···(HX)2复合物体系随着卤素原子序数的递增,结构参数都出现了周期性增加,复合物的相互作用能在整体上呈现减小的总趋势;C6H6···(HCl)2复合物相互作用明显强于C2H2···(HCl)2复合物。
氢键;X-H···π键;结合能
1 引言
乙炔和苯分子分别作为碳碳三键和芳香烃最典型的简单单体和π键电子供体,二者分子中的π键与(HX)2(X=F,Cl,Br,I)相互作用是典型的 X-H…π相互作用。Pierre Carcabal与Dietrich Mootz等对 2-丁炔…HCl,2-丁炔…2HCl所形成的Cl-H…π键的相互作用进行了研究,研究发现二者均能形成稳定的T-Shape构型的几何结构,而且HCl都垂直指向碳碳三键的中点,特别是与2HCl 相互作用时,2HCl 分别从两侧靠近并指向2-丁炔中的C≡C键,形成T-Shape构型的复合物[1-2]。关于芳香π键体系与HX相互作用的研究也相当广泛,而作为芳香 π键体系中最典型也是较简单的单体苯与卤化氢相互作用的研究较多。K.S.Kim等人对苯与卤化氢之间的相互作用进行了比较研究,对苯与卤化氢复合物的几种可能构型进行了比较分析,并优化得到了稳定结构、计算了结合能,研究发现对于具有 C6V对称性的结构,若在较高基组水平下优化将得到与C6轴有倾角的结构,从报道结果显示具有C6V对称性的体系较以HX正对苯环碳碳中点构型、以HX正对苯环顶点构型的结构更稳定,然而,三种结构在高基组水平下优化得到的构型都有一个很小的倾角[3-5]。综上所述,目前对C2H2中π键与卤化氢相互作用形成X-H…π键复合物的在理论和实验上都得到了较广泛的研究。而关于(HF)2、(HCl)2、(HBr)2、(HI)2与乙炔分子 π键相互作用的系统性研究未见报道;因此有必要对乙炔分子π键与(HX)2相互作用的X-H…π键复合物进行深入的研究,弄清楚其相互作用形成复合物的几何结构和相互作用能;其次,关于C2H2、C6H6中的π键与2HX(X=F,Cl,Br,I)相互作用的差异性研究未见报道。
2 实验方法和计算模型
在充分考虑计算量和计算精度的情况下,选用从头算中MP2方法研究复合物的分子间弱相互作用,使用cc- pVTZ(-pp)相关一致基组水平下计算和比较 C2H2、C6H6中的 π键与2HX(X=F,Cl,Br,I)相互作用形成复合物的结构参数和结合能。即在几何结构优化和能量计算过程中对重元素(Br、I)使用同级别的价电子基函数。该论文的工作和数据均是用Gaussian98程序,在贵州省高性能计算化学重点实验室的PC-Linux集群并行计算系统下完成以及在合肥学院化工系分子工程研究室PC机上重复运算和验证后定稿[6]。
构建的乙炔分子中的π键可以在围绕连核的直线与(HX)2相互作用形成含有2个X-H…π键的氢键复合物;苯是最简单的芳香环π单体,也是典型的高度离域的π型电子供体,针对苯环上π电子分布特点,为此构建了C2H2、C6H6与(HX)2形成的复合物体系,如图1所示。
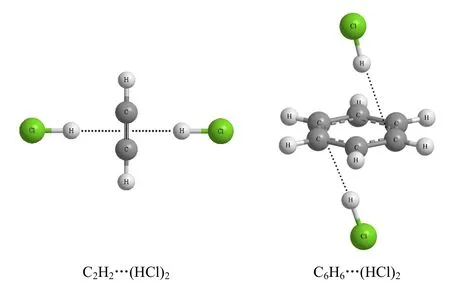
图1 C6H6/C2H2…(HX)2(X=Cl)的模型结构图
3 实验结果与讨论
3.1几何结构
在MP2/cc-pVTZ水平下对(HF)2、(HCl)2与C2H2、C6H6体系分子单体和复合物几何结构进行全自由度能量梯度优化;同理在MP2/cc-pVTZ-pp水平下对(HBr)2、(HI)2与C2H2的所有单体和复合物进行几何结构优化。计算结果如下:
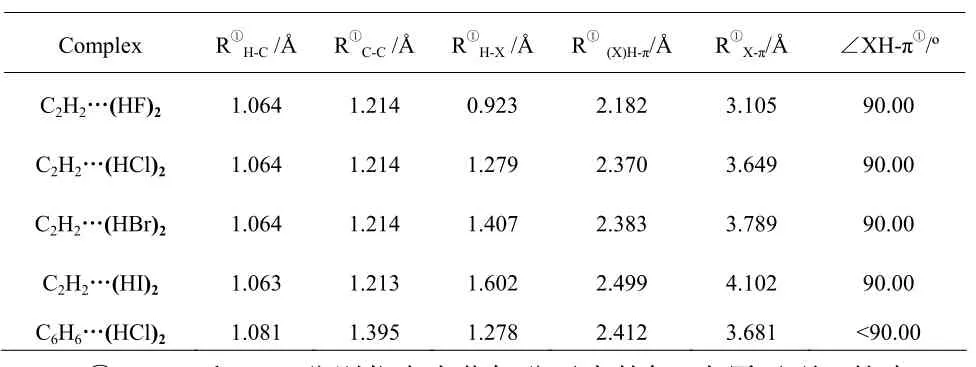
表1 C2H2…(HX)2(X=F,Cl,Br,I)体系在MP2/cc-pVTZ(-pp)水平下优化的结构参数
从表1结构参数可以看出:随着卤元素原子序数的递增,C2H2…(HX)2(X=F,Cl,Br,I)复合物结构参数中,RC-C、RH-C键长几乎没有变化,说明在形成 C2H2…(HX)2复合物对乙炔的结构影响甚微;C2H2…(HX)2中的 RH-X、R(X)H-π、RX···π,数值变化较大,随着卤素原子序数的递增而变大,说明在形成X-H…π键对卤化氢分子的结构影响较大。参数R(X)H-π变大即卤化氢分子到乙炔中C≡C键的距离增加,使X-H…π相互作用减弱,这与表 2中相互作用能参数的计算结果相一致;比较分析表1中 RH-X、R(X)H-π两项数值可以看出,其变化趋势一致,即 C2H2…(HX)2复合物的键长的规律如下:RI-H···π>RBr-H···π>RCl-H···π>RF-H···π,说明了C2H2与(HX)2(X=F,Cl,Br,I)相互作用形成的 X-H…π键键长呈现周期性变化,这主要与(HX)2(X=F,Cl,Br,I)中卤原子半径的增加,卤化氢极化作用的增强,π键的作用距离增加以及X-H…π相互作用减弱相关;而C6H6…(HCl)2复合物中键长参数与C2H2…(HX)2一致。C2H2…(HX)2复合物中键角∠XH-π均为90.00º,即C2H2…(HX)2复合物中X-H与π键的中心在一直线上,HX分子垂直指向π键,C2H2…(HX)2中的 X-H…π键都呈现 T-Shape稳定构型;而C6H6…(HCl)2复合物中X-H与π键所在的面形成的夹角小于90.00º,主要与苯环上的电子高度离域有关。
3.2结合能计算和比较
优化 C2H2…(HX)2(X=F,Cl,Br,I)几何结构和计算其电子密度及Mulliken电荷分布的基础上,在MP2/aug-cc-pVTZ水平下对(HF)2、(HCl)2与C2H2、C6H6的复合物体系进行结合能计算,并用Boys和Bemardi的完全均衡校正法CP校正BSSE;同理在 MP2/aug-cc-pVTZ-pp水平下对(HBr)2、(HI)2与 C2H2所形成的复合物体系进行结合能计算,计算结果如表2所示:
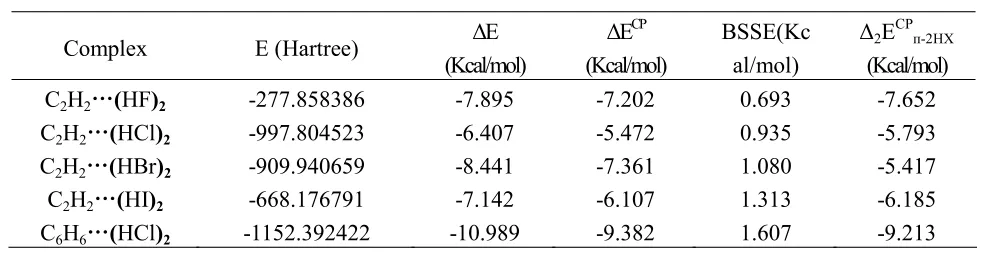
表2 C2H2/C6H6…(HX)2(X=F,Cl,Br,I)在MP2/aug-cc-pVTZ(-pp)水平下能量
从表2中相互作用能ΔECP项可以得出:对于卤化氢单元为(HF)2、(HCl)2时,(HX)2与C2H2相互作用产生较强的结合能,其数值在5.472~7.202Kcal/mol之间,并且相互作用能随着卤元素原子序数的递增而减小;卤化氢单元为(HBr)2、(HI)2时,结合能ΔECP从7.361Kcal/mol减小到6.107Kcal/mol;从相关能校正 BSSE项看,数值随着卤元素原子序数的递增而变大;由于对(HBr)2、(HI)2单元中的Br、I元素使用了价电子基函数,因此出现了C2H2…(HBr)2较C2H2…(HCl)2的结合能大的现象,但从整体相互作用能参数变化趋势上看,ΔE和ΔECP均减小。表2中结合能Δ2ECPп-2HX项可以得出C2H2…(HF)2相互作用能数值最大达到7.652Kcal/mol,说明此时C2H2分子中的π键与(HF)2相互作用最强,而C2H2…(HBr)2相互作用能最小为5.417Kcal/mol,说明此时二者的相互作用最弱。这主要是因为:卤化氢分子中随着卤原子序数的增加,卤化氢的酸性增强,与C2H2中的π键作用距离的明显增加,导致相互作用能的减小,对应的X-H…π相互作用减弱。C6H6…(HCl)2复合物中相互作用能参数ΔE、ΔECP和Δ2ECPп-2HX项均高于复合物的数值,主要与苯环上的电子高度离域、一个苯分子相当含有3个π键,而乙炔分子相当含有2个π键与之作用,因此复合物相互作用明显强于复合物。
4 结论
[1] Carcabal P,Broquier M,Chevalier M,et at.Infrared Spectra of the C2H2-HCl Complexes:An Experimental and Ab initio Study [J].J.Chem.Phys,2000,(113):4876-4883.
[2] Dietrich Mootz and Axel Deeg,2-Butyne and Hydrogen Chloride Cocrystallized:Solid-state Geometry of Cl-H…π Hydrogen Bonding to the Carbon-Carbon Triple Bond [J].J.Am.Chem.Soc.,1992,(114):5887-5888.
[3] Grabowski S J,Leszczynski J.The Enhancement of X-H…πHydrogen Bond by Cooperativity Effects Abinitio and QTAIM Calculations[J].Chem Phys,2009,355(2-3):169-176.
[4] Isabel Rozas,Ibon Alkorta,Jose Elguero.Unusual Hydrogen Bonds:H…πInteractions [J].J.Phys.Chem.A,1997,(101): 9457-9463.
[5] F.A.Baiocchi,J.H.Williams,W.Klemoerer,Molecular Beam Studies of C6F6,C6F3H3,and C6H6, complexes of HF.The Rotational Spectrum of C6H6-HF [J].J.Phys. Chem.,1983, (87):2079-2084.
[6] Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian 98, Revision A [M].Gaussian:Inc,Pittsburgh PA,1998.
Research on the interaction between molecules of C6H6/C2H2···(HX)2
Applying ab initio theoretical MP2 method studies on the complex of C2H2and (HX)2(X=F,Cl,Br,I),which formed by X-H···π bond.They optimized the complexes,then,calculated the BSSE and the binding energy of different systems, analysis the calculation results in the geometry, intermolecular interaction energy parameters, and finally the typical π bond of C6H6molecule and (HCl)2form of X-H···π bond complexes have been studied, composites and compare alkyne to aromatic hydrocarbon two π bond with a hydrogen halide formed by the difference in the nature of hydrogen bonding.The results demonstrated that C2H2···(HX)2,with increasing halogen atomic number,from HF to HI,the bond length have all increasing,however,the binding energy of complex and X-H···π interaction present in the overall decreasing trend, corresponding X-H···π interaction weakened; C6H6···(HCl)2complex interaction was stronger than C2H2···(HCl)2complex.
The Weak Interaction;X-H···π bond;binding energy
061
A
1008-1151(2015)10-0037-02
2015-09-11
国家大学生创新训练计划项目(201411059018,201511059012,201511059013) ; 安徽省自然科学基金(1508085QB31);合肥学院科研发展项目(12KY02ZR)。
阳杰(1984-) ,男,安徽合肥人,合肥学院讲师,硕士,研究方向为分子模拟计算、环保和清洁生产、无机化工及应用。
