酵母穿梭质粒pXZ200的构建
2015-09-10王珊珊徐海洋王世明
王珊珊+徐海洋+王世明


摘要: 利用酿酒酵母高效的同源重组能力,通过PCR扩增技术,将复制子pBBR1片段、KanR(卡纳霉素抗性)片段、酵母复制子2 μ片段这3个两端含有30 bp同源臂的片段电转入酿酒酵母,通过同源重组获得宿主更具广谱性的革兰氏阴性细菌-啤酒酵母穿梭质粒pXZ200。经检测,pXZ200能够在恶臭假单胞杆菌KT2440和大肠杆菌中稳定复制。
关键词: 酿酒酵母;同源重组;穿梭质粒;PCR技术;pXZ200
中图分类号: Q936 文献标志码: A
文章编号:1002-1302(2015)08-0031-03
分子生物学中,利用DNA重组技术使宿主细胞获得某种特殊能力是非常重要而且被广泛利用的方法。然而,这种方法在体外需要经多步的酶切和酶连,很大程度上依赖于载体上限制性核酸内切酶的酶切位点,步骤繁琐,所连接片段一般限于几个kbp大小,很少超过10 kbp,但同源重组的新方法克服了这种限制。
酿酒酵母细胞内有着较其他生物更高效的同源重组修复双链DNA断裂机制,是研究同源重组机制和应用同源重组技术的主要模式生物 [1-2]。在酿酒酵母系统中,15 bp的同源序列就可以介导同源重组的发生,获得重组克隆 [3];当两端的同源序列为30 bp时,有效同源重组的频率可接近80% [4-5]。PCR介导重组技术是指通过人工合成寡核苷酸单链作为PCR引物,用PCR扩增方法得到重组片段的两端带有重组所需同源臂外源的DNA片段 [6-7],是目前应用最为广泛的一种重组技术 [4,8-10],这种方法与传统酶切酶连的基因克隆方法相比,具有很多优点:省去了多步的酶切酶连,操作更为简单快捷;不依赖于限制性酶切位点的存在,不会引入多余的碱基;通过合成特定的同源序列,目的基因可以精确地定位在靶位点上;解决了大片段的基因克隆问题 [11-12]。体外将合成的DNA片段利用酿酒酵母的重组机制,在体内完成多个DNA片段的一步整合已被广泛运用。1970—2008年,DNA合成片段已经从75 bp增加到583 kbp [13]。1970年,Khorana等成功合成丙氨酸转运RNA的完整基因,这是第一次实现化学合成可遗传序列 [14];2008年,Venter研究组合成了由101个DNA片段构建、长度为583 kbp的DNA [15],该DNA通过体外合成25个具有同源臂的DNA片段,然后利用酵母重组机制合成基因组 [16];2009年,Shao等在酵母中一步装配完成含有8个基因的生化途径,含有这个生化途径的重组菌能够利用木糖产生玉米黄质 [17]。
带有完整2μ质粒序列的质粒pXZ198是一个既能在酵母中复制表达、又能在大肠杆菌中复制表达的穿梭质粒,在研究中被广泛应用。但是,pXZ198质粒只含有大肠杆菌的复制子Coli E1,而不能在其他原核细胞中复制表达,而 pBBR1MCS5 却是能够在多种革兰氏阴性菌中稳定复制表达的质粒 [18]。本试验利用酿酒酵母的体内高效重组系统,将广谱复制子pBBR1、卡那霉素抗性基因KanR、酵母复制子2μ序列重组成一新的质粒pXZ200,经验证,该质粒可以在大肠杆菌及恶臭假单胞杆菌KT2440中稳定复制。
1 材料与方法
1.1 材料
1.1.1 主要仪器 恒温培养箱、PCR仪、摇床、低温离心机、冰箱、制冰机、无菌操作台、灭菌锅、电转杯、电转仪、多功能酶标仪等。
1.1.2 主要试剂 DraⅠ限制性内切酶、DNA回收试剂盒、1 mol/L 山梨醇、氨苄抗生素、庆大霉素、卡那霉素、博来霉素等。
1.1.3 主要菌种和质粒 酿酒酵母S288C、大肠杆菌DH5α、恶臭假单胞杆菌KT2440、含有完整2 μ序列及博来霉素抗性基因片段pXZ 的pXZ198质粒、含有KanR片段的pET29a质粒、含有在多种原核细胞中能复制的广谱复制子pBBR1的pBBR1MCS5质粒。
1.1.4 培养基 YPD培养基:2%蛋白胨、1%酵母粉、2%葡萄糖,pH值为7.0,115 ℃灭菌20 min。LB培养基:1%蛋白胨、0.5%酵母粉、1%氯化钠,pH值为7.0,121 ℃灭菌 15 min。固体培养基是在液体培养基的基础上加入1.5%琼脂。
1.2 方法
1.2.1 打靶片段的准备 在-80 ℃接种pXZ198 DH5α至含有100 μg/mL氨苄抗生素、接种pET29a DH5α至含有 50 μg/mL Kan、接种pBBR1MCS5 DH5α至含有30 μg/mL庆大霉素的LB试管中,37 ℃ 200 r/min过夜培养;分别取约 3 mL 菌液,提取质粒 [19]pXZ198、pET29a、pBBR1MCS5,用PCR扩增方法获得两端含有30 bp同源臂的打靶片段,打靶片段PCR对应的引物和模板见表1;每个片段扩增约 120 μL,按照康宁生命科学的试剂盒回收各个片段;回收的各个片段进行0.75%琼脂糖电泳并测各片段的浓度。
1.2.2 感受态细胞的制备 参照文献[17]的方法制作酵母电转感受态、KT2440电转感受态以及采用大肠杆菌DH5α一步法转化感受态细胞 [20]。
1.2.3 电转重组及初步筛选 取50 μL酵母感受态细胞置于预冷的0.2 cm的电转杯中,加入打靶片段pXZ、pBBR1、KanR各5 μL,混匀;擦干电转杯上的水,放入电转化仪的样品槽中,以1.5 kV进行电转化;电转结束,迅速将1 mL已灭菌的YPD培养基加入到电转杯中;在无菌操作台上,将电转杯中液体转移到1.5 mL已灭菌的离心管中,30 ℃ 120 r/min复苏1 h;取100 μL涂布于含有200 μg/mL博来霉素的YPD平板上,共涂10个平板;平板放置于30 ℃恒温培养箱中培养24~36 h。
1.2.4 酵母重组子的验证及pXZ200在酵母中的稳定性 将平板上长出的单菌落随机挑选出3个,并编号为1、2、3,接种至含200 μg/mL博来霉素的YPD试管中,30 ℃ 200 r/min培养24 h,以Kan-R、pBBR1-F为引物对重组子进行PCR鉴定。经PCR鉴定正确的重组子转接至含有200 μg/mL博来霉素的50 mL YPD培养基中,30 ℃ 200 r/min培养24 h以提取质粒。由于酵母菌是真核生物,细胞的破碎难于大肠杆菌。试验采用液氮研磨的方法进行细胞破碎,用酚氯仿反复抽提进行质粒的提取。质粒以pXZR-F、pXZR-R为引物,送至上海桑尼生物科技有限公司测序,测序序列与pBBR1、KanR片段的序列进行比对;测序正确的重组子转接至YPD试管中,30 ℃ 200 r/min过夜培养,反复转接1周并提质粒;对所提质粒进行DraⅠ酶切,酶切体系为10 μL:pXZ200 3 μL、DraⅠ和10×M Bf各1 μL、无菌ddH2O 5 μL;0.75%琼脂糖电泳检测质粒的正确性。
1.2.5 构建质粒在宿主细菌中的复制 将2 μL质粒转入 [21]50 μL DH5α一步法感受态细胞中;转化后加入600 μL的LB复苏1 h;取100 μL涂布于含有50 μg/mL卡那霉素的LB平板上,平板放置于37 ℃恒温培养箱中过夜培养;挑取平板上长出的单菌落,接种到含有50 μg/mL卡那霉素的LB试管中,37 ℃ 200 r/min过夜培养,提取质粒;质粒正确的菌转接至LB试管中,37 ℃ 200 r/min过夜培养,反复转接1周并提取质粒,对所提的质粒进行DraⅠ酶切;0.75%琼脂糖电泳检测质粒的正确性。以同样方法在假单胞杆菌KT2440中检测pXZ200的稳定性。
2 结果与分析
2.1 pXZ200质粒的构建
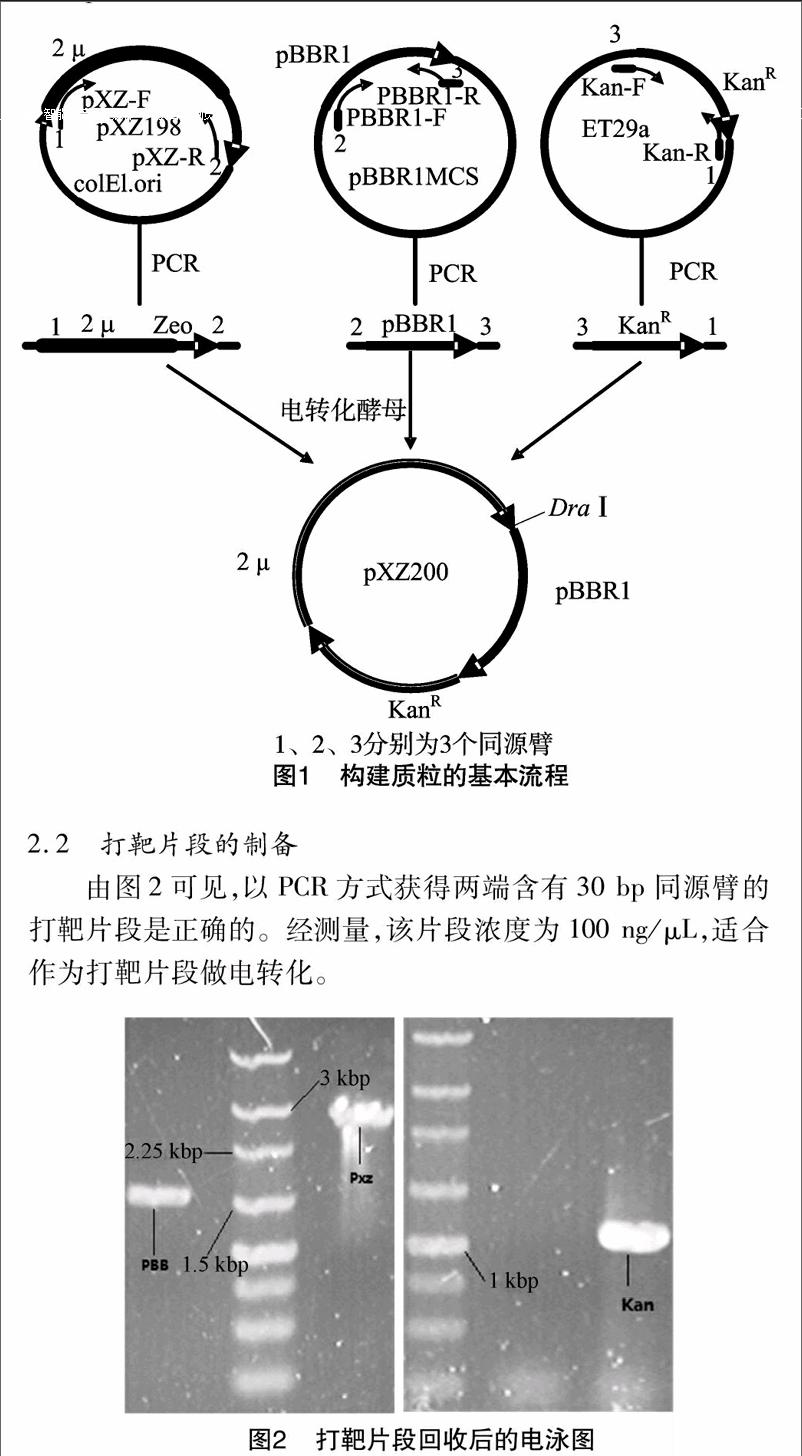
质粒构建的基本流程见图1。
2.2 打靶片段的制备
由图2可见,以PCR方式获得两端含有30 bp同源臂的打靶片段是正确的。经测量,该片段浓度为100 ng/μL,适合作为打靶片段做电转化。
2.3 对重组子的验证及 在酵母中的稳定性。
结果表明,10个含200 μg/mL博来霉素的YPD平板上都长出单菌落,每个平板平均长出20个单菌落;对菌落进行验证统计,重组效率约为70%;对其中3个重组子进行PCR验证,由图3可见,KanR和pBBR1 2个片段的长度大小约为2.5 kbp,1号和2号单菌落是正确的。将2号菌提出的质粒以pXZR-F、pXZR-R为引物测序,经与pBBR1及KanR片段的序列进行比对,符合度达99%,证明该质粒构建正确。
2.4 构建质粒 在假单胞杆菌和大肠杆菌中的表达及稳定性
从酵母菌提取的质粒 转入大肠杆菌和假单胞杆菌KT2440中,提质粒(3 mL菌液)验证都正确(图4)。将含有质粒的大肠杆菌、假单胞杆菌KT2440及验证正确的酵母重组子分别连续转接1周,所提取的质粒经DraⅠ酶切。由图5可见,从大肠杆菌及假单胞杆菌KT2440中提取的质粒和从酵母中提取的质粒相比,大小一致且转接1周后质粒的量没有变化,酶切后质粒大小为5.3 kbp。说明 能够在酵母菌、大肠杆菌DH5α和假单胞杆菌KT2440中稳定复制。
3 结论
传统的质粒构建需要找到一个合适的载体,将目的片段和载体片段在相同的酶切后进行酶连,再转入到宿主菌中鉴定,如果构建的质粒需要插入多个片段,则工作量很大。本试验采用的方法比较简单,只需要在PCR获取目的片段时加入同源臂,然后将所有目的片段混匀,电转到酵母感受态细胞中再进行筛选验证。与传统方法相比,这种方法对多片段质粒的构建简单且易于操作。试验所构建的新质粒 能够在酵母菌及大肠杆菌、假单胞杆菌中稳定复制。
参考文献:
[1] Szostak J W,Orr-Weaver T L,Rothstein R J,et al. The double-strand-break repair model for recombination[J]. Cell,1983,33 (1):25-35.[HJ1.7mm]
[2][JP2]Bhatia P K,Wang Z G,Friedberg E C. DNA repair and transcription[J]. Current Opinion Genetics Development,1996,6(2):146-150.
[3]Judd S R,Petes T D. Physical lengths of meiotic and mitotic gene conversion tracts in Saccharomyces cerevisiae[J]. Genetics,1988,118(3):401-410.
[4]Nickoloff J A,Hoekstra M F. Double-strand break and recombinational repair in Saccharomyces cerevisiae[M]//Nickoloff J A,Hoekstra M F. DNA damage and repair,vol. 1:DNA repair in prokaryotes and lower eukaryotes. Totowa,NJ:Humana Press Inc,1998:335-362.
[5]Willis K K,Klein H L. Intrachromosomal recombination in Saccharomyces cerevisiae:reciprocal exchange in an inverted repeat and associated gene conversion[J]. Genetics,1987,117(4):633-643.
[6][JP3]Kramer K M,Brock J A,Bloom K,et al. Two different types of double- strand breaks in Saccharomyces cerevisiae are repaired by similar RAD52-independent,nonhomologous recombination events[J]. Molecular and Cellular Biology,1994,14(2):1293-1301.
[7]Orr-Weaver T L,Szostak J W. Yeast recombination:the association between double-strand gap repair and crossing-over[J]. Proceedings of the National Academy of Sciences of the United States of America,1983,80(14):4417-4421.
[8][JP2]Schwacha A,Kleckner N. Identification of double Holliday junctions as [JP3]intermediates in meiotic recombination[J]. Cell,1995,83(5):783-791.
[9]Nasmyth K A.Molecular genetics of yeast mating type[J]. Annual Review Genetics 1982,16:439-500.
[10] Thaler D S,Stahl F W. DNA double-chain breaks in recombination of phage lambda and of yeast[J]. Annual Review of Genetics,1988,22:169-197.
[11]Nassif N,Penney J,Pal S,et al. Efficient copying of nonhomologous sequences from ectopic sites via P-element-induced gap repair[J]. Molecular and Cellular Biology,1994,14(3):1613-1625.
[12]Mcgill C,Shafer B,Strathern J. Coconversion of flanking sequences with homothallic switching[J]. Cell,1989,57(3):459-467.
[13]Mueller S,Coleman J R,Wimmer E. Putting synthesis into biology:a viral view of genetic engineering through de novo gene and genome synthesis[J]. Chemistry & Biology,2009,16(3):337-347.
[14] Gibson D G. Synthesis of DNA fragments in yeast by one-step assembly of overlapping oligonucleotides[J]. Nucleic Acids Research,2009,37(20):6984-6990.
[15]Gibson D G,Benders G A,Andrews-Pfannkoch C,et al. Complete chemical synthesis,assembly,and cloning of a mycoplasma genitalium genome[J]. Science,2008,319(5867):1215-1220.
[16]Gibson D G,Benders G A,Axelrod K C,et al. One-step assembly in yeast of 25 overlapping DNA fragments to form a complete synthetic mycoplasma genitalium genome[J]. Proc Natl Acad Sci USA,2008,l05(51):20404-20409.
[17]Shao Z Y,Zhao H,Zhao H M. DNA assembler,an in vivo genetic method for rapid construction of biochemical pathways[J]. Nucleic Acids Research,2009,37(2):e16.
[18]Kovach M E,Elzer P H,Steven Hill D,et al. Four new derivatives of the broad-host-range cloning vector pBBR1MCS,carrying different antibiotic-resistance cassettes[J]. Gene,1995,166 (1):175-176.
[19]任大勇,李 昌,秦艳青,等. 小提质粒快速排除假阳性克隆的新方法[J]. 吉林农业大学学报,2011,33(2):185-188.
[20]涂知明,陈明洁,何光源,等. 三种大肠杆菌高效感受态的制备及转化[J].
