β-内酰胺衍生物的非抗菌生物活性研究进展
2015-07-07吴娟萍
吴娟萍
(湖州市第一人民医院 药剂科,浙江 湖州 313000)
β-内酰胺衍生物的非抗菌生物活性研究进展
吴娟萍Δ
(湖州市第一人民医院 药剂科,浙江 湖州 313000)
β-内酰胺类化合物具有良好的抗菌活性,引起人们的广泛关注。近年来,β-内酰胺类的其他生物活性研究有了长足发展,本文对化合物在抗肿瘤、抗HIV、抗结核、抗寄生虫、抗糖尿病、抗血栓、血脂调节以及治疗神经系统疾病等领域的研究进行简要的综述。
β-内酰胺衍生物;生物活性;抗肿瘤;抗糖尿病
抗生素的发现被认为是20世纪最伟大的科学成就之一,直到现在,β-内酰胺类抗生素,包括青霉素类、头孢菌素类、碳青酶烯类、头霉素类、单环β-内酰胺类等仍是临床应用最广、疗效最好的抗菌药物。20世纪80年代,研究发现一些β-内酰胺类衍生物能够特异性的抑制丝氨酸、半胱氨酸蛋白酶和其他酶类,表现出抗炎、抗病毒、抗癌等多种非抗菌生物活性[1-5],引起了科学家们的极大兴趣,对于β-内酰胺类化合物的其他生物活性研究有了长足发展。现对近年来β-内酰胺衍生物(见图1)在抗肿瘤、抗HIV、抗结核、抗寄生虫、抗糖尿病、抗血栓、神经系统疾病治疗以及降血脂等领域的研究进行综述。
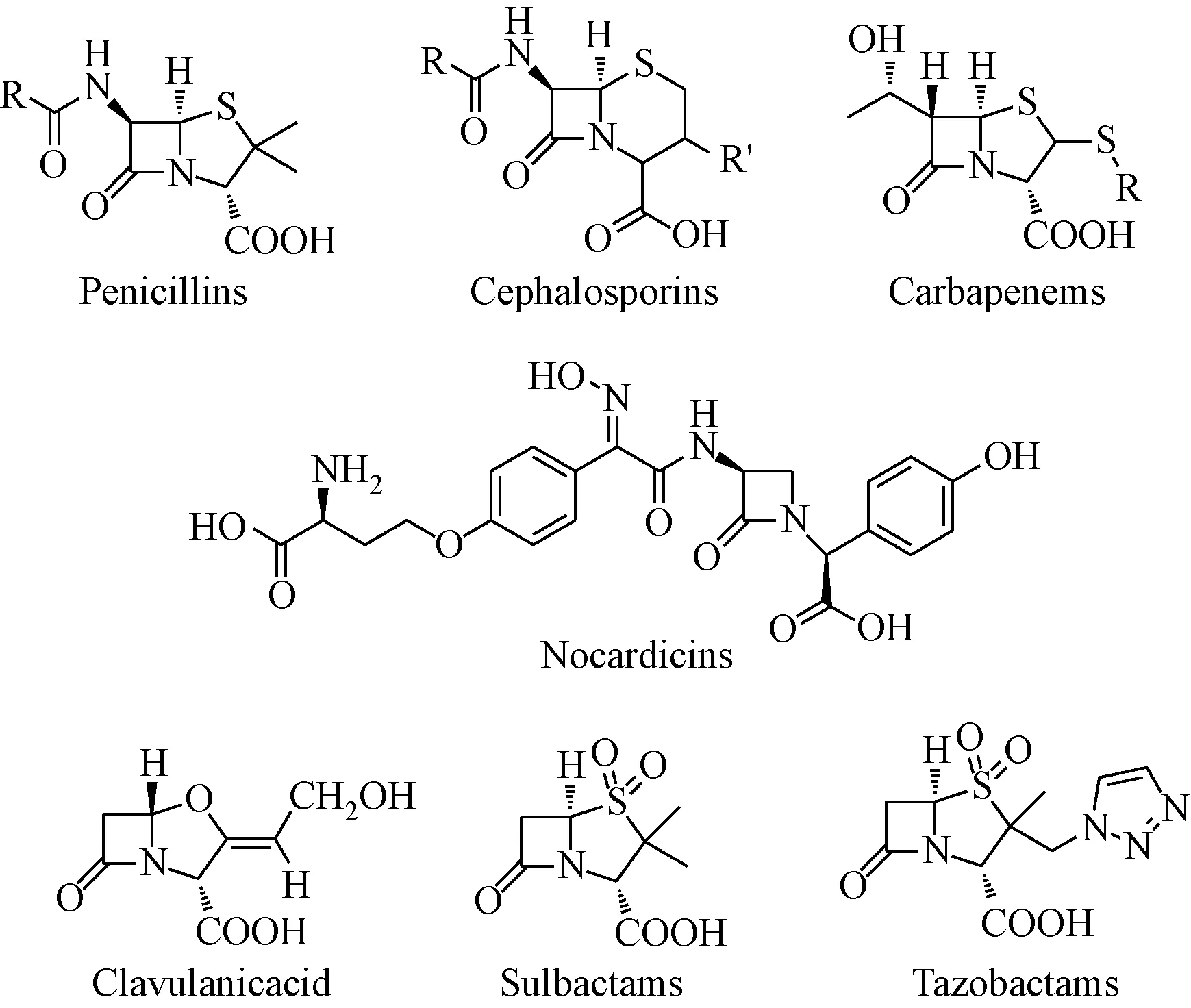
图1 β-内酰胺类抗生素Fig.1 β-lactam derivatives
1 β-内酰胺衍生物的生物活性
β-内酰胺又称氮杂环丁烷-2-酮,是最常见的四元内酰胺,见图2。β-内酰胺与其它杂环并和或N-1,C-3和C-4位被不同的取代基取代表现出多样的生物学活性。
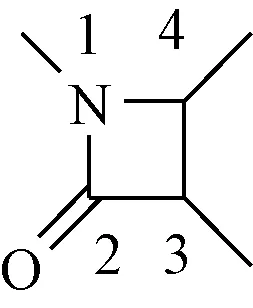
图2 β-内酰胺的氮杂环丁烷-2-酮结构Fig.2 Structure of azetidine-2-one in β-lactam derivatives
1.1 抗肿瘤活性
1.1.1 PARP抑制剂:Turos等[6-7]在研究N-硫代单环β-内酰胺(见图3)的抗菌活性时合成了化合物1-1、1-2和1-3,发现其能激活肿瘤细胞凋亡通路而不影响正常细胞。研究表明[8],这些化合物特异性的剪切恶性肿瘤细胞内的PARP(多腺苷二磷酸核糖聚合酶),能抑制多种肿瘤细胞株的增殖。化合物1-1和1-2对乳腺癌移植瘤小鼠的肿瘤生长抑制率分别为49%和18%,且没有明显的毒副作用。
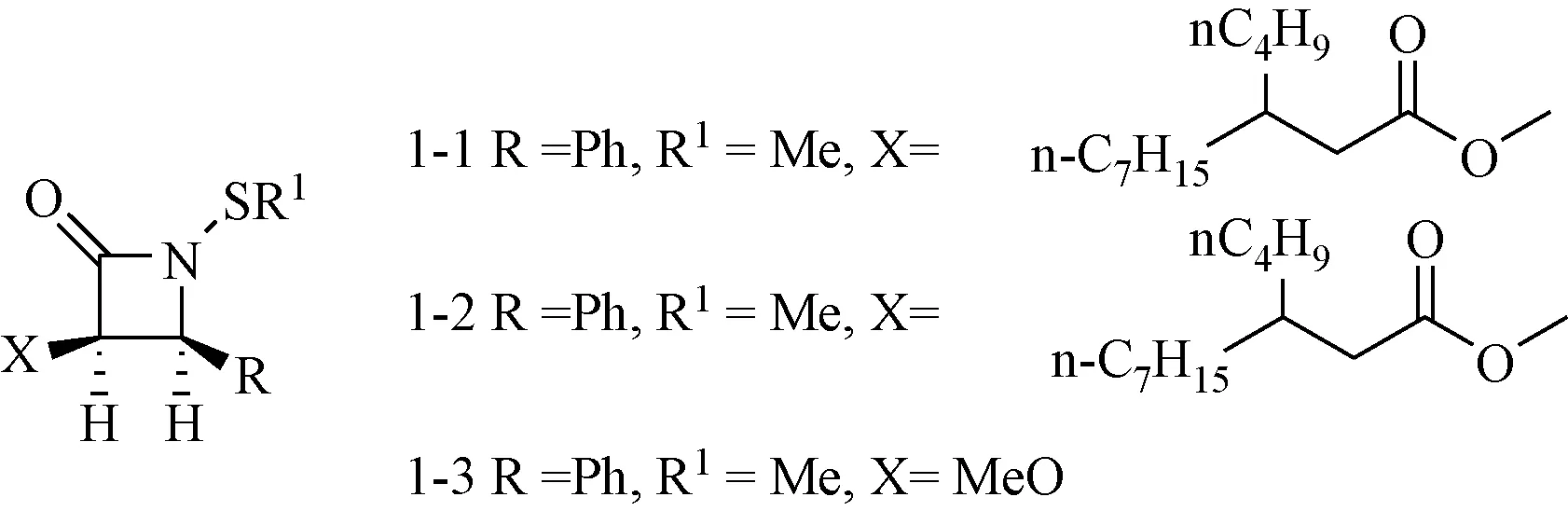
图3 N-硫代单环β-内酰胺衍生物Fig.3 N-thiolated monocyclic β-lactam derivatives
Turos等[6]提出,1-1、1-2和1-3的作用机制可能与N-硫代烷基单环β-内酰胺中存在3个可被亲核进攻的位点有关。见图4。
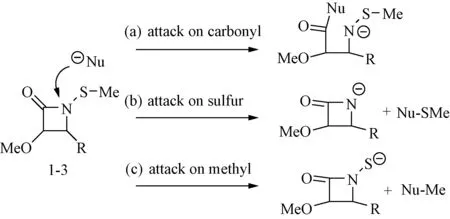
图4 化合物1-3的作用机制Fig.4 Mechanism of compound 1-3
1.1.2 基质金属蛋白酶抑制剂:基质金属蛋白酶(matrix metalloproteinase,MMPs)是一类活性依赖于锌离子的蛋白水解酶,参与细胞外基质(extracellular matrix,ECM)的降解和组织重塑。MMPs对基底膜和细胞外基质的降解为肿瘤细胞的浸润和转移创造了有利条件。在肿瘤细胞中,尤其是恶性肿瘤中,MMPs的表达增高,活性增强,因此基质金属蛋白酶抑制剂可以用于抗肿瘤治疗。
2002年,Cainelli等[9]设计合成了一系列新型4-亚烷基β-内酰胺类人白细胞弹性蛋白酶(human leukocyte elastase,HLE)和哺乳动物明胶酶抑制剂(见图5),发现部分化合物(1-4~1-11)具有一定的基质金属蛋白酶抑制活性。其中化合物1-4,1-5,1-6在200 μM浓度下对MMP-2的抑制率分别为53%、65%、56%。这是β-内酰胺化合物首次被报道用于MMPs抑制剂的研究。
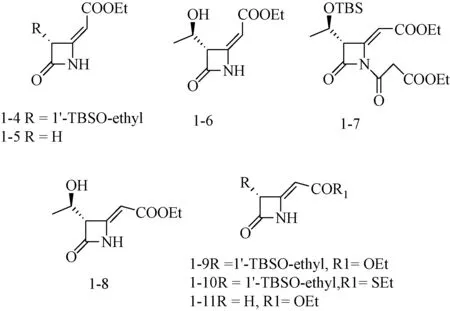
图5 4-亚烷基β-内酰胺类Fig.5 4-alkylene β-lactam derivatives
Cainelli等[10]进一步对这些化合物的β-内酰胺的N-1,C-3和C-4位进行衍生化(见图6),发现引入没食子酰基可以大大提高该类化合物的活性,N-1位没食子酰基衍生物1-12,C-3位侧链上的羟基与没食子酸、没食子酸苄基醚成酯衍生物1-13、1-14对MMP-2的抑制率分别提高到81%、92%和61%(100μM)。
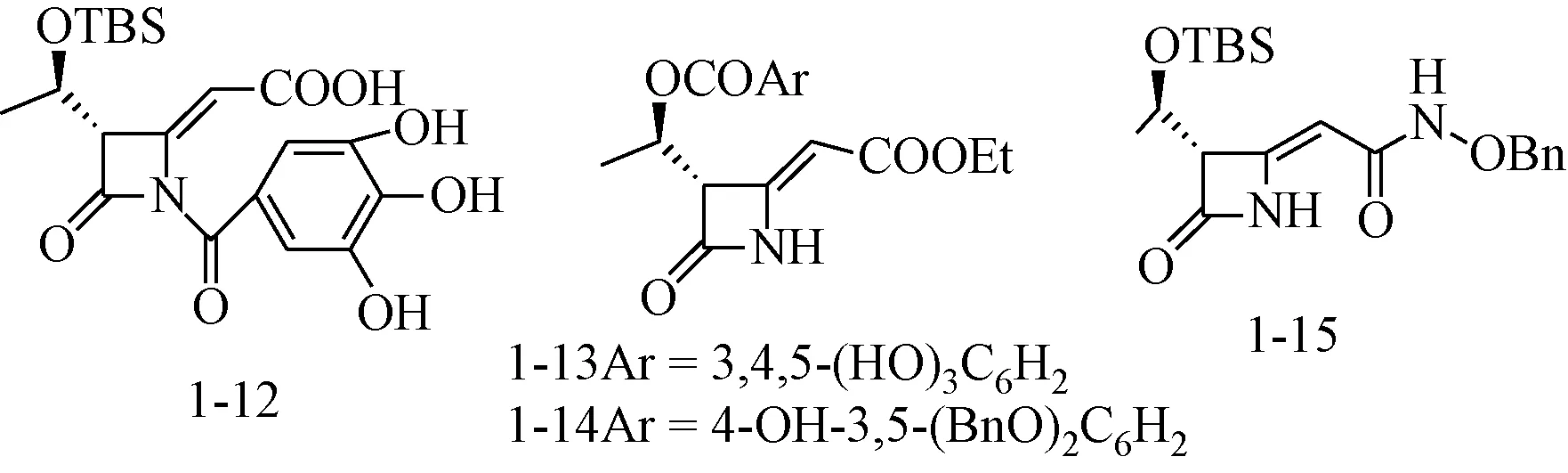
图6 没食子酰基β-内酰胺衍生物Fig.6 Galloyl β-lactam derivatives
1.1.3 人类20S蛋白酶体抑制剂:泛素-蛋白酶体系统参与体内蛋白质的降解,具有清除损伤、衰老和错误折叠蛋白的功能。蛋白酶体抑制剂通过抑制蛋白酶体的活性阻断了泛素-蛋白酶体通路,造成细胞内蛋白降解异常,诱导细胞凋亡,具有广泛的抗肿瘤活性。
化合物1-16是由诺华公司开发的非共价蛋白酶体抑制剂,其IC50为15nM[11]。研究人员在1-16的C-端引入β-内酰胺,合成了一类新型的“自杀型“蛋白酶体抑制剂1-17~1-23(见图7)。对其构效关系的研究表明,β-内酰胺C-3位是R构型是维持活性的重要条件,当C-3位为S构型时其活性相比R构型下降约1000倍。体外肿瘤细胞株增殖抑制活性筛选结果显示,化合物1-19、1-23对乳腺癌细胞株MDA-MB435表现出了显著的抗增殖活性,IC50分别为34 nM和32 nM。LC-MS分析结果表明,化合物1-17可与蛋白酶体的β5亚基共价结合,发挥”自杀性抑制“活性[12]。见表1。

图7 引入β-内酰胺的新型“自杀型”蛋白酶体抑制剂Fig.7 Novel suicide proteasome inhibitor by introducingβ-lactam表1 1-17~1-23对乳腺癌细胞20S蛋白酶体的抑制活性Tab.1 Inhibitory activity of compound 1-17 to 1-23 on 20S proteasome from human breast cancer
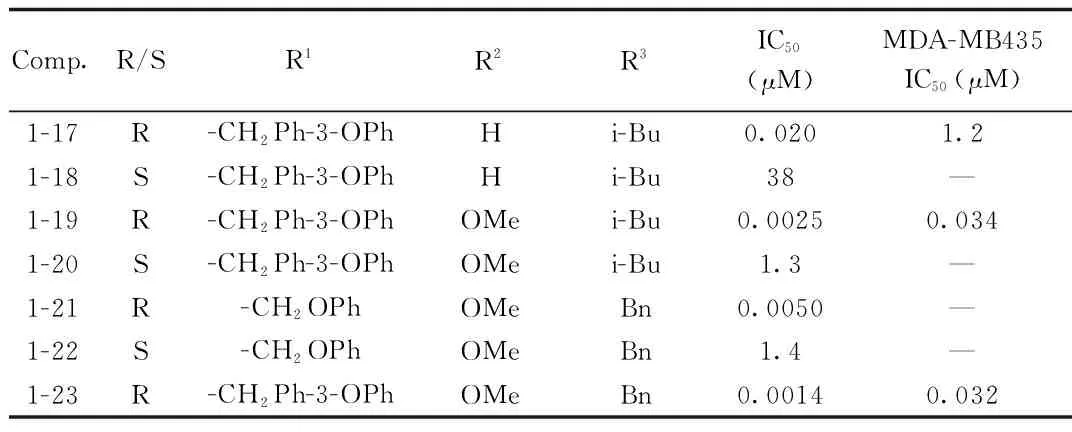
Comp.R/SR1R2R3IC50(μM)MDA-MB435IC50(μM)1-17R-CH2Ph-3-OPhHi-Bu0.0201.21-18S-CH2Ph-3-OPhHi-Bu38—1-19R-CH2Ph-3-OPhOMei-Bu0.00250.0341-20S-CH2Ph-3-OPhOMei-Bu1.3—1-21R-CH2OPhOMeBn0.0050—1-22S-CH2OPhOMeBn1.4—1-23R-CH2Ph-3-OPhOMeBn0.00140.032
1.1.4 DNA嵌入剂:含有碱性侧链的多环芳香族化合物被认为能够嵌入DNA,发挥抗肿瘤活性。Becker等将多环芳香片段稠二萘与N-甲基哌嗪经丁二酰胺连接,合成了化合物1-24,1-24对MCF-7、MDA-231、OVCAR、PC-3、HL-60、HT-29等肿瘤细胞株表现出了一定的增殖抑制活性,验证了上述假说[13]。在此基础上,他们将开链的二酰胺片段环合,设计合成了构象限制的β-内酰胺化合物1-25。见图8。在体外抗肿瘤活性筛选中,化合物1-25对人乳腺癌MCF-7细胞株表现出了比1-24(IC50为40.0 μM)和阳性对照顺铂(IC50为10.05 μM)更优越的抑制活性,IC50为9.91 μM[14]。
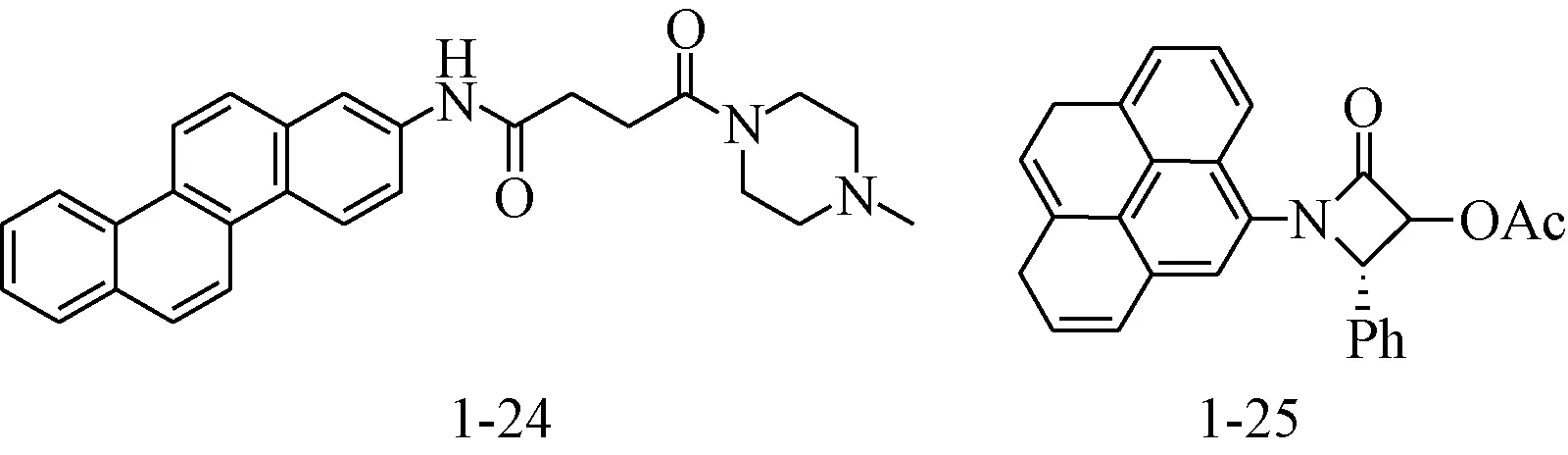
图8 DNA嵌入剂Fig.8 DNA intercalator
1.1.5 微管蛋白抑制剂:考布他汀(CA-4),是从南非矮柳树树皮中分离出的多羟基二苯乙烯类化合物,能竞争性的结合微管蛋白秋水仙碱结合位点,抑制肿瘤细胞有丝分裂过程中纺锤体的形成,对多种肿瘤细胞包括包括多药耐药细胞株具有增殖抑制活性[15-16]。CA-4P是将CA-4的羟基磷酸酯化的前体药物,其水溶性和靶向选择性均优于CA-4,目前已经处于临床三期[17]。但CA-4及其衍生物在贮存和使用过程中易发生双键构型的变构,导致活性大大降低,针对这一缺点,Farida Tripodi等用β-内酰胺取代双键合成了一系列构象限制的1, 4-二芳基-2-氮杂环丁酮化合物(见图9)。其中 (±)-trans-1-26、(±)-tans-1-27表现出了优秀的细胞毒活性,对十二指肠腺癌细胞的增殖抑制IC50为3~15nM。该类化合物不仅可以抑制微管蛋白聚合,使细胞有丝分裂停止在G2/M期,同时激活了腺苷酸活化的蛋白激酶(AMPK)和半胱氨酸蛋白酶-3,诱导了肿瘤细胞的凋亡[18]。
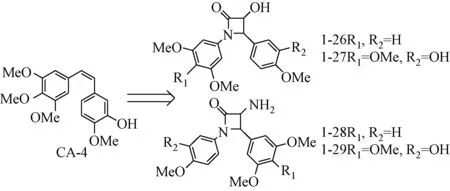
图9 1, 4-二芳基-2-氮杂环丁酮化合物Fig.9 1,4-diaryl-2-azetidinone
1.2 抗HIV活性 HIV-1蛋白酶是天冬氨酸类蛋白水解酶,它催化HIV-gap和gap-pol基因编码产生的前体多聚蛋白水解,形成病毒成熟必需的酶类和结构性蛋白,使病毒具有感染性。HIV-1蛋白酶抑制剂可抑制这一过程,使病毒繁殖停留在不成熟、无感染性病毒颗粒阶段,达到抑制病毒复制的目的[19-21]。
Sperka等[22]采用高通量筛选的办法,对126个单环β-内酰胺衍生物进行了抗HIV活性筛选,发现化合物1-30(见图10)对HIV-1蛋白酶的抑制活性达97%,进一步研究表明其抑制方式为非竞争性抑制。
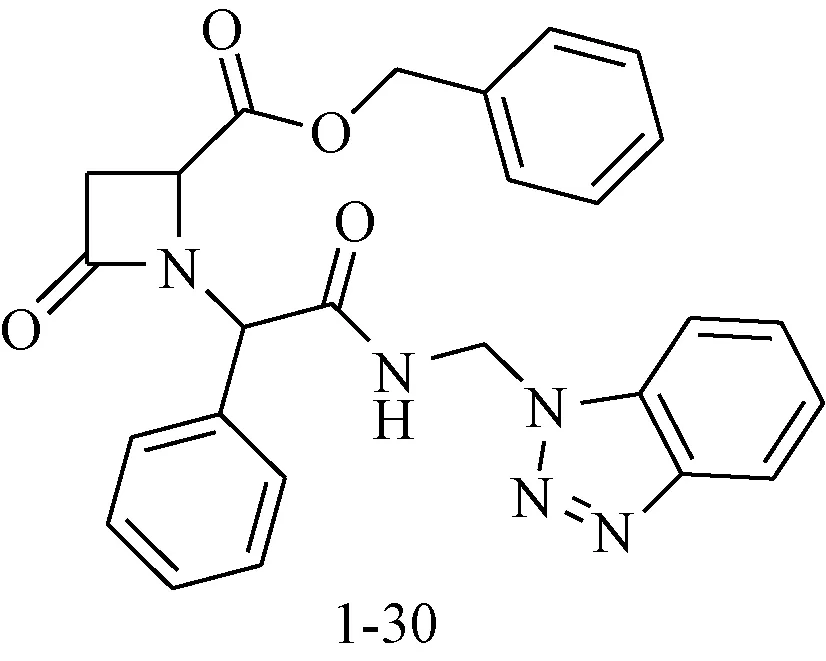
图10 抗HIV活性化合物1-30Fig.10 Anti-HIV agents 1-30
1.3 抗结核活性 肺结核在全世界仍然有着较高的死亡率,2011年全球有大约870万人死于肺结核。结核分枝杆菌易发生突变,对异烟肼和利福平等一线抗结核药物产生多药耐药性,多重耐药结核病已经成为了控制结核病传播的一大挑战[23],寻找具有全新结构或新的作用模式的新型抗结核药物有着重要意义。
Vashi等[24]以4-亚硝基百里酚为原料合成了一系列氮杂环丁烷-2-酮衍生物,并测试了其抗菌和抑制结核生长活性,化合物 1-31~1-33表现出对结核杆菌较高的抑制活性。
Ilango等[25]合成了一系列新型4-芳基-3-氯-N-(3,4,5-三羟基-苯甲酰胺基)-2-氮杂环丁酮化合物。在采用微孔板Alamar蓝方法测定该系列化合物对人型结核分枝杆菌的抑制活性的试验中,化合物1-34~1-37的最小抑菌浓度(minimal inhibitory concentration,MIC)与异烟肼相当,分别为0.76, 0.57, 0.83和0.62 μg/mL,异烟肼为0.56 μg/mL。见图11。
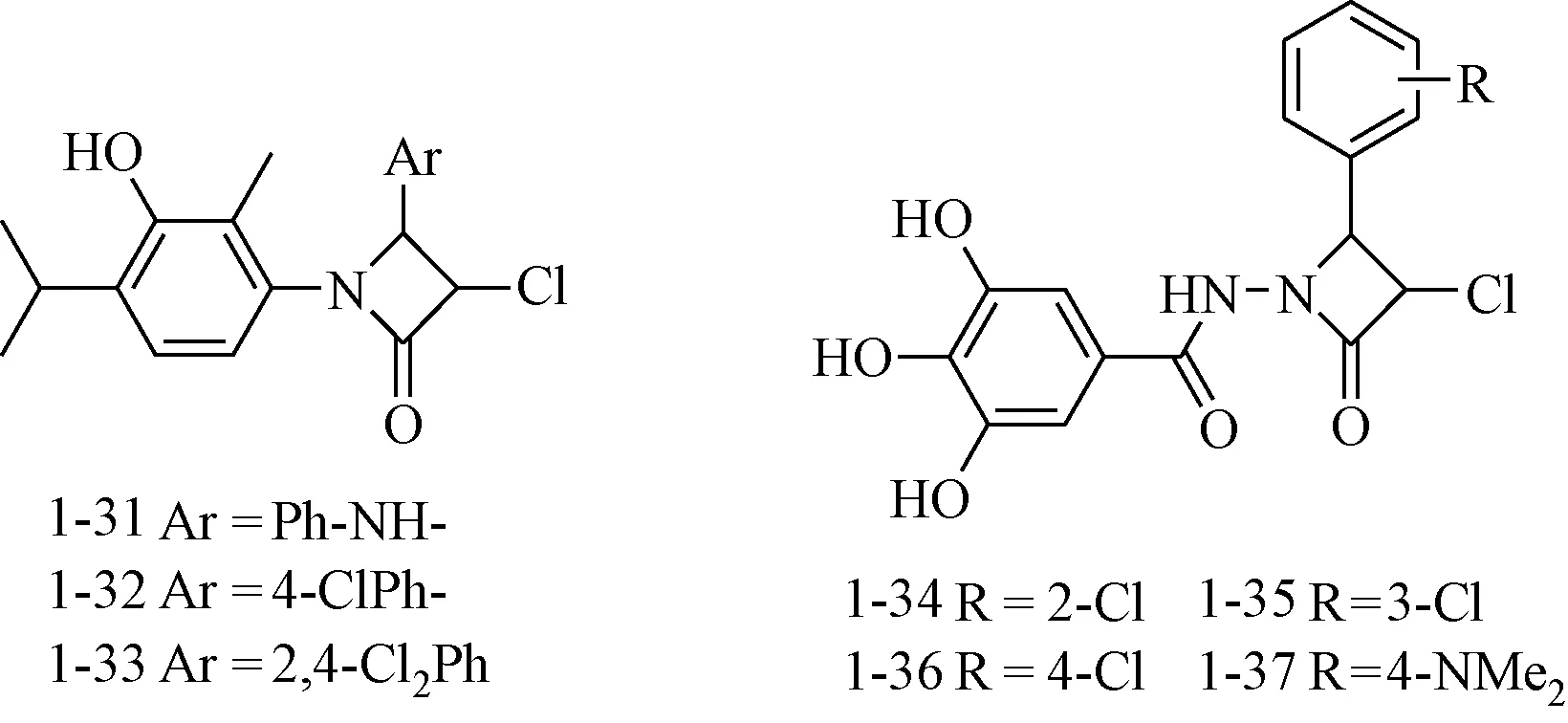
图11 抗结核活性化合物Fig.11 antituberculosis agents
1.4 利什曼原虫D-甘露糖磷酸化转移酶抑制剂 利什曼虫可以导致多种人类疾病,其在沙蝇载体和人类宿主的寄生能力与其分泌特殊的多糖磷酸化分子(PGs)密切相关,D-甘露糖磷酸化转移酶(eMPT)在PGs的合成中发挥着关键作用,可以作为有效的抗利什曼原虫病药物作用靶点[26-29]。
Ruhela等[30]以PG结构中的重复单体1-38为先导物,合成了一系列1-38的β-内酰胺衍生物。其中化合物1-39及其N-烷基化衍生物1-40在浓度为0.6 mM和0.2 mM时,对D-甘露糖磷酸化转移酶(eMPT)的抑制率为50%(见图12)。

图12 利什曼原虫D-甘露糖磷酸化转移酶抑制剂Fig.12 Inhibitor of eMPT in leishmania
1.5 抗糖尿病活性 过氧化物酶体增殖剂激活受体(peroxisome proliferators-activated receptors,PPAR)是细胞核激素受体超家族的一员, 包括PPARα,PPAR β/δ,PPARγ 3种亚型。PPARα受体,主要在肝脏中表达,与脂肪酸氧化和脂质代谢有关;PPARγ受体主要在脂肪和组织和巨噬细胞中表达,激活PPARγ可以促进前脂肪细胞分化,增加外周组织胰岛素敏感性,因此PPARα/γ双重受体激动剂可以用来治疗糖尿病和糖尿病引发的血脂异常e。
Muraglitazar是默克和施贵宝默克公司合作开发的PPARα/γ双通道激动剂[32],目前处于临床三期用于治疗2型糖尿病(见图13)。
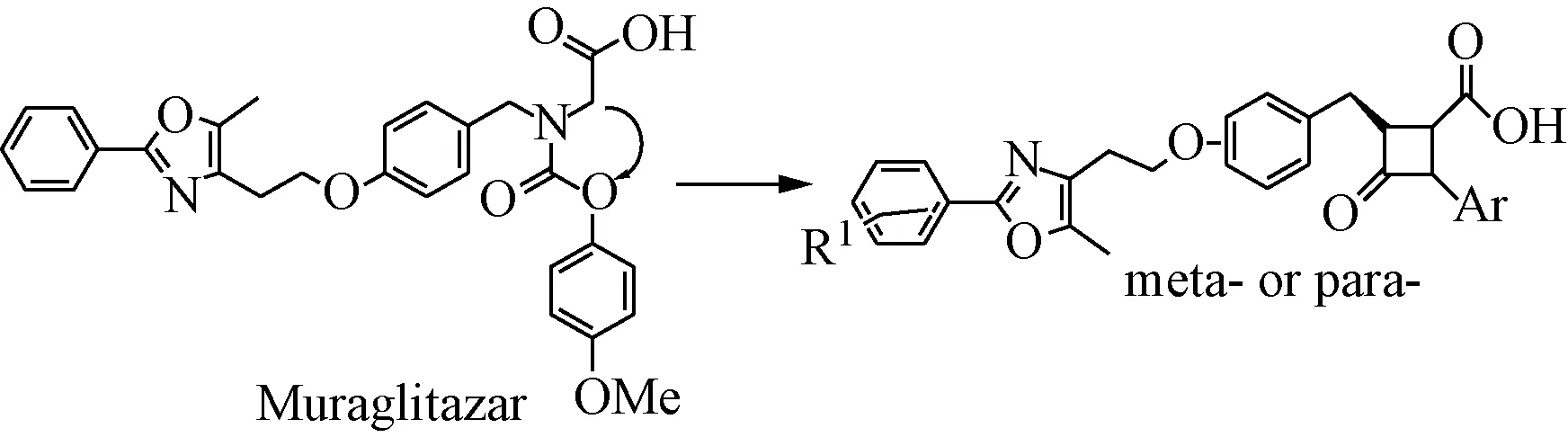
图13 PPARα/γ双通道激动剂MuraglitazarFig.13 Muraglitazar of PPARα/γ dual agonist
Wang等[33]以Muraglitazar为先导物,将其末端的氨基乙酸成环进行构象限制,并对N,O原子的位置进行转换合成了一系列新型β-内酰胺类PPARα/γ 双重受体激动剂。构效关系研究表明,顺式(3S, 4S)构型活性优于(3S, 4R)反式异构体,N-1位4-甲氧基苯基取代衍生物1-41表现出了强效的PPARα/γ激动活性(PPAR α/γ EC50=70/90 nM),能够显著降低血糖水平(43%)。见图14。
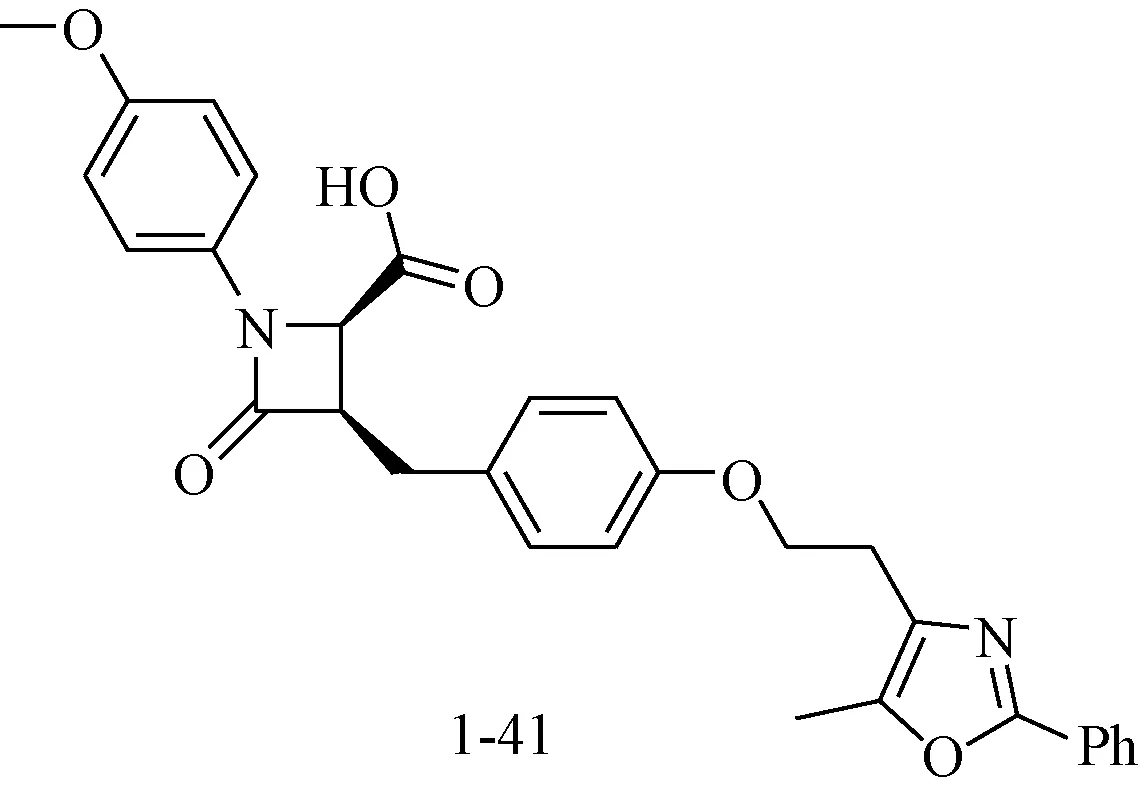
图14 新型β-内酰胺类PPARα/γ双重受体激动剂Fig.14 Novel β-lactams of PPARα/γ dual agonist
1.6 凝血酶抑制活性 凝血酶是一种丝氨酸蛋白酶,在凝血级联反应中由凝血因子Xa激活凝血酶原生成。凝血酶的主要作用是水解可溶性纤维蛋白原使其形成不溶性纤维蛋白,促进血小板聚集,使血液凝固,抑制凝血酶可用于治疗动脉和静脉血栓[34-35]。
文献报道β-内酰胺衍生物(如1-42、1-43)是有效的丝氨酸蛋白酶抑制剂,提示β-内酰胺结构可用于凝血酶抑制剂的开发。在此基础上,Han等在β-内酰胺的C-3位连接精氨酸侧链合成了一系列β-内酰胺凝血酶抑制剂,大多数化合物表现出了较好的凝血酶抑制活性,其中化合物1-44活性最好,IC50为2nM[36]。见图15。

图15 凝血酶抑制剂Fig.15 Thrombin inhibitors
1.7 抗利尿激素V1a受体拮抗活性 精氨酸抗利尿激素(arginine vasopressin,AVP)是由中枢神经系统的神经分泌细胞特异性分泌的环状九肽激素,可与中枢神经系统和周缘组织中的特定GPCRs受体结合发挥广泛的生理作用,目前被确定的有3个受体亚型——V1a,V1b和V2受体。V1a受体亚型主要分布在大脑特别是大脑皮层、边缘系统、下丘脑和脑干中,选择性V1a受体拮抗剂可发挥枢神经系统疾病(如精神疾病、认知障碍等)治疗作用[37-39]。
Guillon等[40]在高通量筛选选择性V1a受体拮抗剂过程中发现了化合物LY307174,该化合物对人抗利尿激素V1a受体有较好的拮抗活性,IC50=45nM,以LY307174为先导,一系列β内酰胺衍生物被设计和合成用于V1a受体拮抗剂的筛选,其中化合物SRX246和SRX251对V1a受体表现出了很强的亲和力,Ki值分别为0.3和0.66 nM。大鼠口服给药后,其脑内药物Cmax为28 ng/mL和48 ng/mL,相当于28 nM和62 nM,是其Ki值的100倍。见图16。
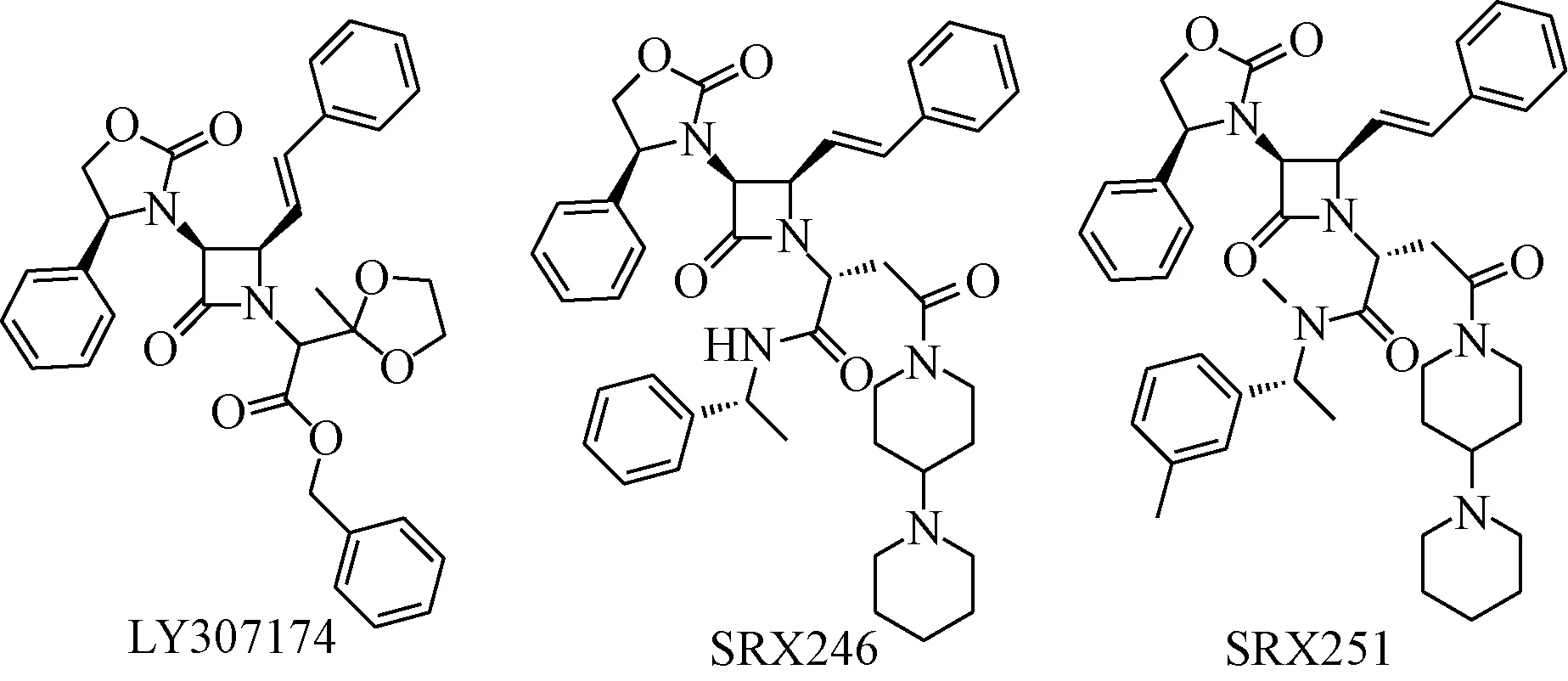
图16抗利尿激素V1a受体拮抗剂Fig.16 Receptor antagonist of vasopressin V1a
1.8 胆固醇吸收抑制活性 依折麦布是先灵葆雅公司20世纪90年代开发的新型2-氮杂环丁酮类胆固醇吸收抑制剂(见图17),它可以抑制小肠上皮细胞对肠道中胆固醇的吸收,从而降低了血浆和肝脏中胆固醇含量。依折麦布2002年首次在德国上市,用于治疗家族性高胆固醇血症、原发性高胆固醇血症和植物甾醇血症,是世界上首个能选择性抑制胆固醇吸收的药物。
1.8.1 依折麦布的发现过程:2-氮杂环丁酮类胆固醇吸收抑制剂的发现源于对酰基辅酶A胆固醇酰基转移酶(acyl coenzyme A-cholesterol acyltransferase,ACAT)抑制剂的研究[41]。在ACAT作用下,体内的胆固醇形成胆固醇酯,促进了食物中胆固醇的吸收和肝脏中脂蛋白的合成。化合物1-45是当时已知的ACAT抑制剂,但其只能降低高胆固醇膳食喂养的仓鼠的肝脏胆固醇酯(CE)水平,而不能降低其血清胆固醇(SE)含量[42-43]。
Vaccaro等[44]对1-45进行构象限制合成了二氢化茚类化合物1-46,1-46的体内和体外活性都有所提高,与此同时,Burnett等[45]提出了氮杂环丁烷衍生物1-47。见图18。
1-47是由亚胺与苯乙酸乙酯缩合得中间体1-48,再由硝酸铈铵(CAN)氧化脱保护、还原、酰化而得。在1-48的合成过程中得到了副产物1-49。体外及体内的活性测试结果显示1-47、1-48、1-49都不是潜在的ACAT抑制剂,但是副产物1-49表现出了一定的降低血清胆固醇的活性(-26% @ 100 mg/kg)。受此启发,Burnett合成了一系列的氮杂环丁酮衍生物,构效关系的研究表明化合物SCH48461表现出了突出的降低高胆固醇膳食喂养的仓鼠的肝脏胆固醇酯(-93% @ 10mg /kg)和血清胆固醇水平(-43% @ 10 mg/kg)的效果[46-47]。见图19。
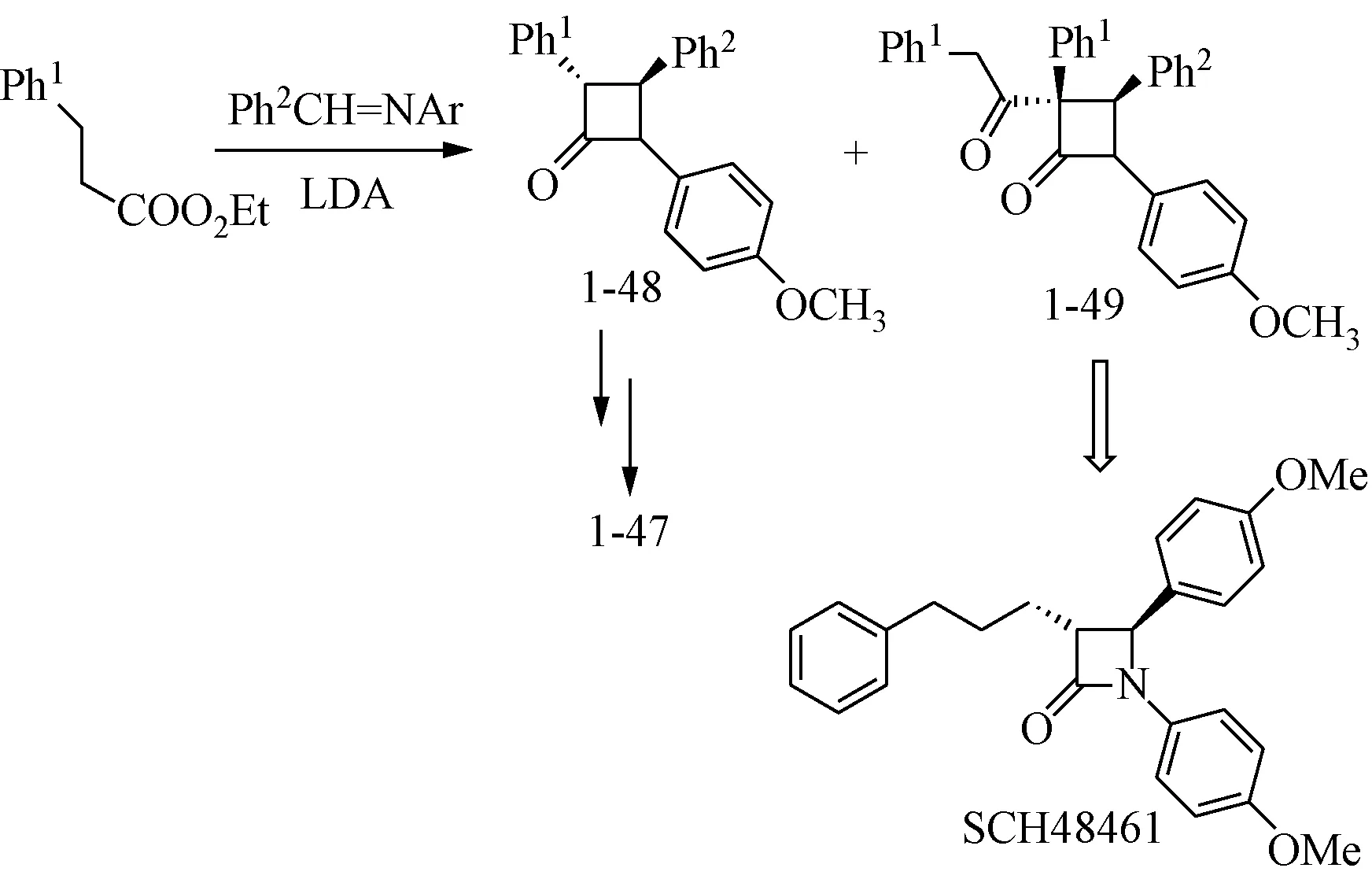
图19 SCH48461发现过程Fig.19 Discovery process of SCH48461
Heek等[48]在对SCH48461及其代谢产物进行药理研究时发现,SCH48461本身活性较弱,主要是其代谢产物发挥着降低血清胆固醇作用。Kvaernφ等[49]对SCH48461的代谢产物进行了分离确证,并合成了其可能代谢产物,体内活性测试表明,SCH48461的R2位去甲基化代谢产物1-51、R3位羟基化代谢产物1-52的降胆固醇活性大大降低,而C-3位侧链苄位S构型羟基化、C-4位4-甲氧基苯基去甲基化代谢后则可以显著的提高降胆固醇活性,化合物1-55的ED50为0.3 mg/kg,其体内活性约是SCH48461的10倍。见图20、表1。
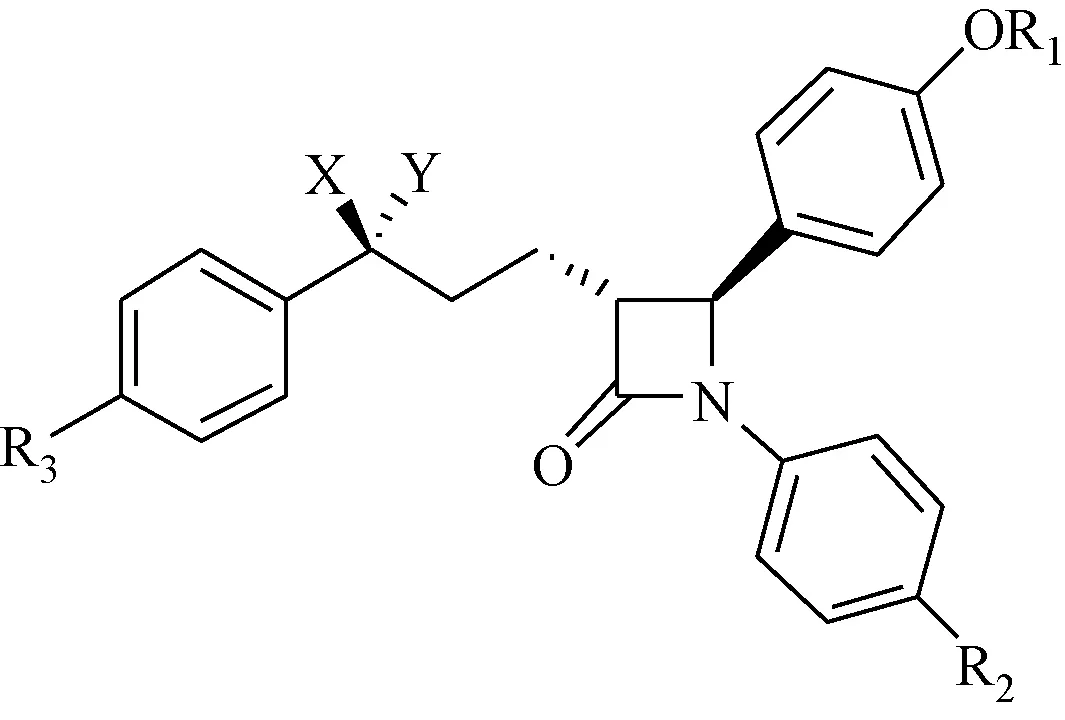
图20 SCH48461及其代谢产物Fig.20 SCH48461 and its metabolites表2 SCH48461及其代谢产物的体内活性Tab.2 Activity of SCH48461 and its metabolites in vivo
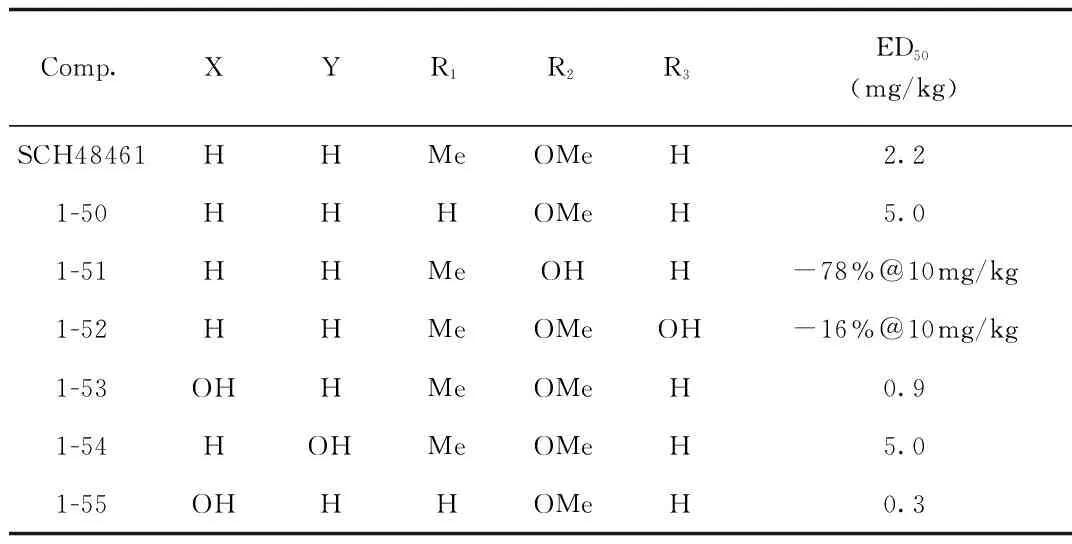
Comp.XYR1R2R3ED50(mg/kg)SCH48461HHMeOMeH2.21-50HHHOMeH5.01-51HHMeOHH-78%@10mg/kg1-52HHMeOMeOH-16%@10mg/kg1-53OHHMeOMeH0.91-54HOHMeOMeH5.01-55OHHHOMeH0.3
基于此研究,Rosenblum等[50]在化合物1-55的R3位和R2位引入氟原子阻断了该位点在体内的不利代谢途径合成了SCH58235,即后来的依折麦布。从表1.3可以看出依折麦布对于高胆固醇膳食喂养的仓鼠、大鼠、恒河猴以及狗的降低血清胆固醇的疗效均显著优于SCH48461。见图21、表3。
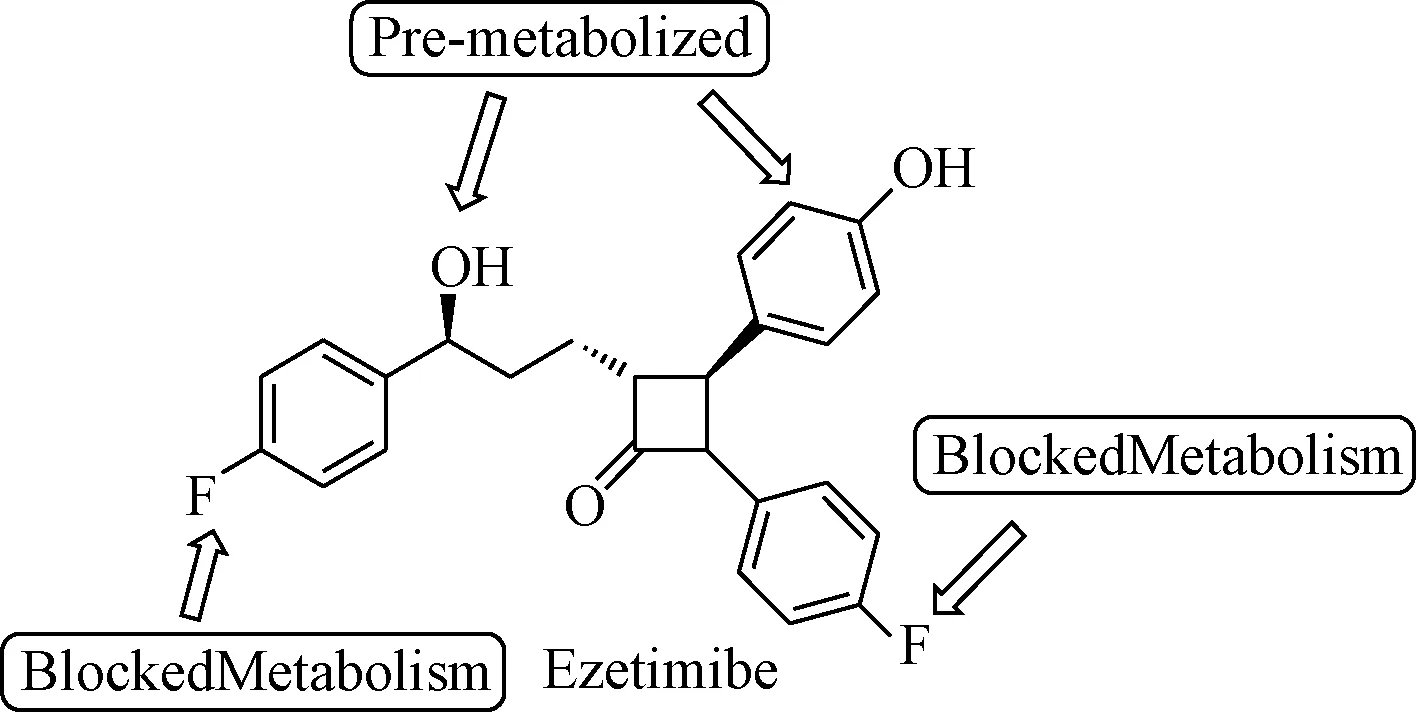
图21 抗体内不利代谢的依折麦布Fig.21 Ezetimibe of antimetabolite in vivo表3 依折麦布与SCH48461的体内活性比较Tab.3 Comparison of activity of ezetimibe and SCH48461 in vivo
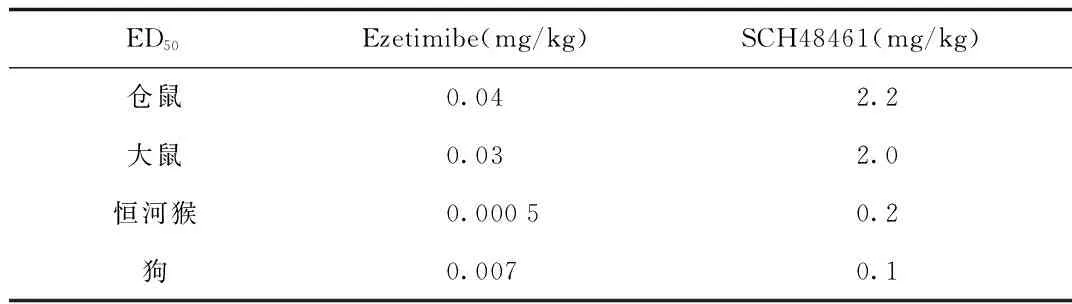
ED50Ezetimibe(mg/kg)SCH48461(mg/kg)仓鼠0.042.2大鼠0.032.0恒河猴0.00050.2狗0.0070.1
2 小结
近年来,随着越来越多的β-内酰胺衍生物被合成和研究,β-内酰胺广泛的活跃于化学和医药领域。本文综述了近年来β-内酰胺在抗肿瘤、抗HIV、抗结核、抗寄生虫、抗糖尿病、抗血栓、神经系统疾病治疗以及降血脂等方面的应用,以及β-内酰胺的化学合成方法的进展。根据这些研究,我们相信β-内酰胺衍物有着多样的生物活性和广阔的药用前景,未来将会有更多的β-内酰胺衍生物被发现和用于各类疾病的治疗。
[1] Veinberg G,Vorona M,Shestakova I,et al.Design of beta-lactams with mechanism based nonantibacterial activities[J].Curr Med Chem,2003,10(17):1741-1757.
[2] Xing BG,Rao JH,Liu RR.Novel beta-lactam antibiotics derivatives: Their new applications as gene reporters,antitumor prodrugs and enzyme inhibitors[J].Mini Rev Med Chem,2008,8(5):455-471.
[3] Mehta PD,Sengar NP,Pathak AK.2-Azetidinone-A new profile of various pharmacological activities[J].Eur J Med Chem,2010,45(12):5541-5560.
[4] Singh GS.beta-lactams in the new millennium.Part-Ⅱ: Cephems,oxacephems,penams and sulbactam[J].Mini Rev Med Chem,2004,4(1):93-109.
[5] Singh GS.beta-lactams in the new millennium.Part-I: Monobactams and carbapenems[J].Mini Rev Med Chem,2004,4(1):69-92.
[6] Kuhn D,Coates C,Daniel K,et al.N-thiolated bicyclic and monocyclic beta-lactams[J].Front Biosci,2004,9:2605-2617.
[7] Ren XF,Konaklieva MI,Shi HC,et al.Studies on nonconventionally fused bicyclic beta-lactams[J].J Org Chem,1998,63:8898-8917.
[8] Frezza M,Garay J,Chen D,et al.Induction of tumor cell apoptosis by a novel class of N-thiolated beta-lactam antibiotics with structural modifications at N-1 and C-3 of the lactam ring[J].Int J Mol Med,2008,21(6):689-695.
[9] Cainelli G,Galletti P,Garbisa S,et al.4-alkylidene-azetidin-2-ones: Novel inhibitors of leukocyte elastase and gelatinase[J].Bioorg Med Chem,2003,11(24):5391-5399.
[10] Cainelli G,Galletti P,Garbisa S,et al.4-alkyliden-beta-lactams conjugated to polyphenols: Synthesis and inhibitory activity[J].Bioorg Med Chem,2005,13(22):6120-6132.
[11] Furet P,Imbach P,Noorani M,et al.Entry into a new class of potent proteasome inhibitors having high antiproliferative activity by structure-based design[J].J Med Chem,2004,47(20):4810-4813.
[12] Imbach P,Lang M,García-Echeverría C,et al.Novel beta-lactam derivatives: potent and selective inhibitors of the chymotrypsin-like activity of the human 20S proteasome[J].Bioorg Med Chem Lett,2007,17(2):358-362.
[13] Becker FF,Banik BK.Polycyclic aromatic compounds as anticancer agents: Synthesis and biological evaluation of some chrysene derivatives[J].Bioorg Med Chem Lett,1998,8(20):2877-2880.
[14] Banik I,Becker FF,Banik BK.Stereoselective synthesis of beta-lactams with polyaromatic imines: Entry to new and novel anticancer agents[J].J Med Chem,2003,46(1):12-15.
[15] Lin CM,Ho HH,Pettit GR,et al.Antimitotic Natural-Products Combretastatin-a-4 and Combretastatin-a-2-Studies on the Mechanism of Their Inhibition of the Binding of Colchicine to Tubulin[J].Biochemistry,1989,28(17):6984-6991.
[16] Nam NH.Combretastatin A-4 analogues as antimitotic antitumor agents[J].Curr Med Chem,2003,10(17):1697-1722.
[17] West CM,Price P.Combretastatin A4 phosphate[J].Anticancer Drugs,2004,15(3):179-187.
[18] Tripodi F,Pagliarin R,Fumagalli G,et al.Synthesis and Biological Evaluation of 1,4-Diaryl-2-azetidinones as Specific Anticancer Agents: Activation of Adenosine Monophosphate Activated Protein Kinase and Induction of Apoptosis[J].J Med Chem,2012,55(5):2112-2124.
[19] Tözsér J.Stages of HIV replication and targets for therapeutic intervention[J].Curr Top Med Chem,2003,3(13):1447-1457.
[20] Tözsér J,Oroszlan S.Proteolytic events of HIV-1 replication as targets for therapeutic intervention[J].Curr Pharm Des,2003,9(22):1803-1815.
[21] Randolph JT,DeGoey DA.Peptidomimetic inhibitors of HIV protease[J].Curr Top Med Chem,2004,4(10):1079-1095.
[22] Sperka T,Pitlik J,Bagossi P,et al.Beta-lactam compounds as apparently uncompetitive inhibitors of HIV-1 protease[J].Bioorg Med Chem Lett,2005,15(12):3086-3090.
[23] Johnston JC,Shahidi NC,Sadatsafavi M,et al.Treatment Outcomes of Multidrug-Resistant Tuberculosis: A Systematic Review and Meta-Analysis[J].PLoS One,2009,4(9):e6914.
[24] Vashi BS,Mehta DS,Shah VH.Synthesis and Biological-Activity of 4-Thiazolidinones,2-Azetidinones,4-Imidazolinone Derivatives Having Thymol Moiety[J].Indian J Chem,Section B: Organic Chem Including MEd Chem,1995,34:802-808.
[25] Ilango K,Arunkumar S.Synthesis,Antimicrobial and Antitubercular Activities of Some Novel Trihydroxy Benzamido Azetidin-2-one Derivatives[J].Trop J Pharm Res,2011,10:219-229.
[26] Descoteaux A,Turco SJ.Functional aspects of the Leishmania donovani lipophosphoglycan during macrophage infection[J].Microbes Infect,2002,4(9):975-981.
[27] Turco SJ,Descoteaux A.The lipophosphoglycan of Leishmania parasites[J].Annu Rev Microbiol,1992,46:65-94.
[28] McConville MJ,Ferguson MA.The structure,biosynthesis and function of glycosylated phosphatidylinositols in the parasitic protozoa and higher eukaryotes[J].Biochem J,1993,294(Pt 2):305-324.
[29] Späth GF,Lye LF,Segawa H,et al.Persistence without pathology in phosphoglycan-deficient Leishmania major[J].Science,2003,301(5637):1241-1243.
[30] Ruhela D,Chatterjee P,Vishwakarma RA.1-Oxabicyclic beta-lactams as new inhibitors of elongating MPT-a key enzyme responsible for assembly of cell-surface phosphoglycans of Leishmania parasite[J].Org Biomol Chem,2005,3(6):1043-1048.
[31] Gervois P,Fruchart JC,Staels B.Drug Insight: mechanisms of action and therapeutic applications for agonists of peroxisome proliferator-activated receptors[J].Nat Clin Pract Endocrinol Metab,2007,3(2):145-156.
[32] Devasthale PV,Chen S,Jeon Y,et al.Design and synthesis of N-[(4-methoxyphenoxy)carbonyl]-N-[[4-[2-(5-methyl-2-phenyl-4-oxazolyl)ethoxy]phenyl]methyl]glycine [Muraglitazar/BMS-298585],a novel peroxisome proliferator-activated receptor alpha/gamma dual agonist with efficacious glucose and lipid-lowering activities[J].J Med Chem,2005,48:2248-2250.
[33] Wang W,Devasthale P,Farrelly D,et al.Discovery of azetidinone acids as conformationally-constrained dual PPAR alpha/gamma agonists[J].Bioorg Med Chem Lett,2008,18(6):1939-1944.
[34] Royle,N.J.; Clarkson,R.; Wong,Z.; Jeffreys,A.J.,Preferential Localization of Hypervariable Minisatellites near Human Telomeres[J].Cytogenetics and Cell Genetics 1987,46,685-686.
[35] Degen SJ,Davie EW.Nucleotide sequence of the gene for human prothrombin[J].Biochemistry,1987,26(19):6165-6177.
[36] Han WT,Trehan AK,Wright JJ,et al.Azetidin-2-one derivatives as inhibitors of thrombin[J].Bioorg Med Chem,1995,3(8):1123-1143.
[37] Ring RH.The central vasopressinergic system: examining the opportunities for psychiatric drug development[J].Curr Pharm Des,2005,11(2):205-225.
[38] Landgraf R.The involvement of the vasopressin system in stress-related disorders[J].CNS Neurol Disord Drug Targets,2006,5(2):167-179.
[39] Landgraf R.The brain vasopressin system is a final common pathway in trait anxiety and depression-like behavior[J].Neuropeptides,2006,40:142-143.
[40] Guillon CD,Koppel GA,Brownstein MJ,et al.Azetidinones as vasopressin V1a antagonists[J].Bioorg Med Chem,2007,15(5):2054-2080.
[41] Sliskovic DR,Picard JA,Krause BR.ACAT inhibitors: the search for a novel and effective treatment of hypercholesterolemia and atherosclerosis[J].Prog Med Chem,2002,39:121-171.
[42] Clader JW,Berger JG,Burrier RE,et al.Substituted (1,2-Diarylethyl)Amide Acyl-Coa-Cholesterol Acyltransferase Inhibitors-Effect of Polar Groups on in-Vitro and in-Vivo Activity[J].J Med Chem,1995,38(10):1600-1607.
[43] Heinonen TM.Acyl coenzyme A: cholesterol acyltransferase inhibition: potential atherosclerosis therapy or springboard for other discoveries?[J].Expert Opin Investig Drugs,2002,11(11):1519-1527.
[44] Vaccaro W,Amore C,Berger J,et al.Inhibitors of acyl CoA:cholesterol acyltransferase[J].J Med Chem,1996,39(8):1704-1719.
[45] Burnett DA,Caplen MA,Davis HR Jr,et al.2-Azetidinones as Inhibitors of Cholesterol Absorption[J].J Med Chem,1994,37(12):1733-1736.
[46] Clader JW,Burnett DA,Caplen MA,et al.2-Azetidinone cholesterol absorption inhibitors: structure-activity relationships on the heterocyclic nucleus[J].J Med Chem,1996,39(19):3684-3693.
[47] Salisbury BG,Davis HR,Burrier RE,et al.Hypocholesterolemic Activity of a Novel Inhibitor of Cholesterol Absorption,Sch-48461[J].Atherosclerosis,1995,115(1):45-63.
[48] Van Heek M,France CF,Compton DS,et al.In vivo metabolism-based discovery of a potent cholesterol absorption inhibitor,SCH58235,in the rat and rhesus monkey through the identification of the active metabolites of SCH48461[J].J Pharmacol Exp Ther,1997,283(1):157-163.
[49] Kvaernφ L,Werder M,Hauser H,et al.Synthesis and in vitro evaluation of inhibitors of intestinal cholesterol absorption[J].J Med Chem,2005,48(19):6035-6053.
[50] Rosenblum SB,Huynh T,Afonso A,et al.Discovery of 1-(4-fluorophenyl)-(3R)-[3-(4-fluorophenyl)-(3S)-hydroxypropyl]-(4S)-(4-hydroxyphenyl)-2-azetidinone (SCH 58235): a designed, potent, orally active inhibitor of cholesterol absorption[J].J Med Chem,1998,41(6):973-980.
(编校:王俨俨)
Progress of bioactivities other than antibacterial of β-lactam derivatives
WU Juan-pingΔ
(Department of Pharmacy, The First People’s Hospital of Huzhou, Huzhou 313000, China)
β-lactam compounds exhibit good antibacterial activity, which aroused widespread attention. Recently, the researches on the biological activities of beta-lactam derivatives have made great development. In this paper, we will review the recent research progress of β-lactam derivatives in the field of anti-tumor, anti-HIV, anti-tuberculosis, anti-parasitic, anti-diabetes, antithrombotic, plasma lipids regulation and treatment on nervous system diseases.
β-lactam derivatives; biological activity; anti-tumor; anti-diabetes
吴娟萍,通信作者,女,本科,副主任药师,研究方向:抗菌药物,E-mail:hulsph@sina.cn。
R914
A
1005-1678(2015)10-0161-07
