超高效合相色谱法定性与定量分析补肾健脑颗粒
2015-07-02朱瑞娟王胡芳李灿柳小亚封士兰周围
朱瑞娟王 波 胡芳 李灿柳小亚封士兰*周围*
1(兰州大学药学院,兰州 730000) 2(甘肃出入境检验检疫局综合技术中心,兰州 730000)
超高效合相色谱法定性与定量分析补肾健脑颗粒
朱瑞娟1王 波2胡芳1李灿1柳小亚1封士兰*1周围*2
1(兰州大学药学院,兰州 730000)2(甘肃出入境检验检疫局综合技术中心,兰州 730000)
用超高效合相色谱法(Ultra performance convergence chromatography,UPC2)建立补肾健脑颗粒及各药材提取物的指纹图谱,对补肾健脑颗粒主要色谱峰进行归属分析,并测定有效成分β-蜕皮甾酮和松果菊苷的含量。样品经乙醇提取后,进样1 μL,用Waters ACQUITY UPC2TMBEH柱(100 mm×3.0 mm,1.7 μm)分离,柱温为40℃,以超临界CO2-0.05%H3PO4-甲醇溶液为流动相,梯度洗脱,流速为0.8 mL/min。分析补肾健脑颗粒和各药材提取物的UPC2指纹图谱,利用各峰相对保留时间、紫外光谱图及部分对照品归属主峰。结果表明,补肾健脑颗粒UPC2指纹图谱中15个主峰来源明确,其中12号峰为β-蜕皮甾酮,含量为380 μg/g, 15号峰为松果菊苷,含量为9.562 mg/g。与HPLC和UPLC法相比,本方法简便、快速,精密度高,重现性好。
超高效合相色谱;补肾健脑颗粒;β-蜕皮甾酮;松果菊苷
1 引 言
补肾健脑胶囊临床上主要用于治疗儿童多动症,且疗效显著[1]。但该制剂存在工艺简单粗糙,服用量大,质量难以控制,小儿服用顺应性不好的缺点,并且没有定性定量方法控制其质量。因此本研究在保证疗效的基础上将补肾健脑胶囊制成颗粒剂,克服了其服用困难的缺陷,并采用色谱法对该制剂进行定性与定量分析,以控制其质量。补肾健脑颗粒是由肉苁蓉、怀牛膝、远志、石菖蒲等药材经过提取、浓缩、干燥、粉碎、制粒工序制成的复方制剂。本研究用HPLC法和UPLC法分析补肾健脑颗粒,在多次优化色谱条件后,都不能将β-蜕皮甾酮和松果菊苷分离。因此,采用一种新的分离技术—超高效合相色谱(Ultra performance convergence chromatography,UPC2)[2,3],建立了补肾健脑颗粒及其组成药材的指纹图谱,归属分析了补肾健脑颗粒指纹图谱中主要色谱峰,并建立β-蜕皮甾酮和松果菊苷的含量测定方法,该方法能将β-蜕皮甾酮和松果菊苷有效分离,与HPLC和UPLC方法相比,更简便、快速,且分离度高,重复性好。本实验利用UPC2定性定量分析了补肾健脑颗粒,在复杂中药制剂质量控制方面提供了新的思路。
2 实验部分
2.1 仪器与试剂
2695高效液相色谱仪、2996PDA检测器(Waters公司);ACQUITY超高效液相色谱仪、ACQ-PDA检测器(Waters公司);ACQUITY超高效合相色谱仪、ACQ-UPC2-PDA检测器(Waters公司);KQ-400DB超声仪(昆山市超声仪器有限公司);EYELAOSB-2100旋转蒸发仪(日本东京理化)。
松果菊苷(MUST-13072403)、β-蜕皮甾酮(MUST-12122111),均购自上海顺勃生物技术有限公司;乙腈(色谱纯,德国莫克公司);H3PO4(分析纯,北京化工厂);甲醇(分析纯,天津北方天医化学试剂厂);甲醇(色谱纯,山东禹王试剂公司);纯净水(兰州大学第一医院生产);补肾健脑颗粒(三批中试样品,由兰州中医院脑病康复医院提供);肉苁蓉、怀牛膝、远志、石菖蒲药材由兰州黄河药市提供。
2.2 色谱条件
采用Acqtity UPC2BEH色谱柱;系统背压为1800 psi;色谱柱温度为40℃;柱流速为0.8 mL/min;进样量为1 μL;流动相分别为超临界CO2(流动相A)和0.05%H3PO4-甲醇溶液(流动相B),梯度洗脱:0~0.3 min,99%A;0.3~4 min,99%~90%A;4~16 min,90%~70%A;16~18 min,70%~60%A; 18~20 min,60%A;20~21 min,60%~99%A;21~25 min,99%A。检测波长248 nm。
2.3 对照品溶液的制备
称取松果菊苷对照品16.65 mg,加50%甲醇溶解并定容至25 mL棕色容量瓶中,得666 mg/L的松果菊苷对照品溶液。称取β-蜕皮甾酮对照品1.62 mg,用甲醇溶解并定容至50 mL,得32.4 mg/L β-蜕皮甾酮对照品溶液。
2.4 样品溶液的制备
补肾健脑颗粒制备:按处方比例称取肉苁蓉、远志、怀牛膝、石菖蒲等适量,煎煮,收集滤液、浓缩、干燥、粉碎、加适量辅料制粒得补肾健脑颗粒。阴性样品制备:按上述方法分别制备缺肉苁蓉和缺怀牛膝的阴性样品。
药材提取物的制备:按处方分别称取各药材适量,按照处方提取工艺提取,收集滤液,浓缩,干燥,得药材提取物。
2.5 供试品溶液的制备
分别取补肾健脑颗粒约1.00 g,各药材提取物约0.50 g,精密称定,置于50 mL圆底烧瓶中,加90%乙醇25 mL,回流提取2次,每次1 h,合并滤液,回收乙醇,残渣用甲醇溶解定容至25 mL,用0.22 μm微孔滤膜滤过,取续滤液,即得。
3 结果与讨论
3.1 提取溶剂及提取条件的选择
本研究旨在尽可能多且完全地提取样品中的有效成分,根据各药材所含主要有效成分的性质,本实验分别以50%甲醇、60%甲醇、70%甲醇、甲醇、60%乙醇、70%乙醇、80%乙醇、90%乙醇对补肾健脑颗粒及各药材提取物进行超声和回流提取,结果表明,90%乙醇回流提取,提取物指纹图谱中峰最多。又考察提取时间和提取次数对提取效率(峰面积)的影响,结果表明,25 mL溶剂提取两次,每次30 min,提取效率最高。
3.2 色谱条件的选择
考察了色谱柱对样品分离效果的影响,分别选用ACQUITY UPC2TMHSS C18SB柱和ACQUITY UPC2TMBEH柱进样分析,结果表明,在本实验条件下ACQUITY UPC2TMBEH柱对样品峰有较好的分离效果。由于流动相对分离效果影响很大,本实验分别用超临界CO2-甲醇、超临界CO2-0.05%H3PO4-甲醇、超临界CO2-乙腈、超临界CO2-0.05%H3PO4-乙腈梯度洗脱,结果表明,有机溶剂采用甲醇时,样品峰有较好的分离,但有些峰峰形不好,稍有拖尾、峰形展宽。向有机溶剂中加入少量H3PO4,配成0.05%H3PO4-甲醇溶液,能改善峰形展宽及拖尾现象。因此选择超临界CO2-0.05%H3PO4-甲醇作为流动相。
3.3 紫外检测波长的选择
对样品进样分析,全波长扫描。结果248 nm波长下的指纹图谱样品峰数最多,且吸收相对较强,易于主峰归属研究。因此最终选择248 nm作为检测波长。
3.4 方法学考察
3.4.1 专属性考察取供试品溶液、对照品溶液、阴性供试品溶液,按2.2节进样检测,比较阴性样品色谱图与对照品和样品色谱图(图1),在松果菊苷和β-蜕皮甾酮峰吸收位置无干扰,表明本方法专属性好。
3.4.2 线性考察取松果菊苷对照品溶液1.00,1.25,1.67,2.50,5.00和10.00 mL,置于10 mL棕色容量瓶中,用50%甲醇溶解并定容,得到质量浓度分别为66.60,83.25,111.00,166.50,333.00和666.00 mg/L的系列溶液。按照上述色谱条件,每个浓度依次进样1 μL,以浓度(mg/L)为横坐标,以峰面积为纵坐标,进行线性回归。取β-蜕皮甾酮对照品溶液1.00,1.25,1.67,2.50,5.00和10.00 mL,置于10 mL容量瓶中,用甲醇溶解并定容,得到质量浓度分别为 3.24,4.05,5.40,8.10,16.20和32.40 mg/L的系列溶液。同样色谱条件下,每个浓度依次进样1 μL,以浓度(mg/L)为横坐标,以峰面积为纵坐标,进行线性回归(表1)。补肾健脑颗粒在16min内得到良好分离,在一定浓度范围内,松果菊苷和β-蜕皮甾酮呈现良好的线性关系(r>0.9996),精密度RSD<2%。
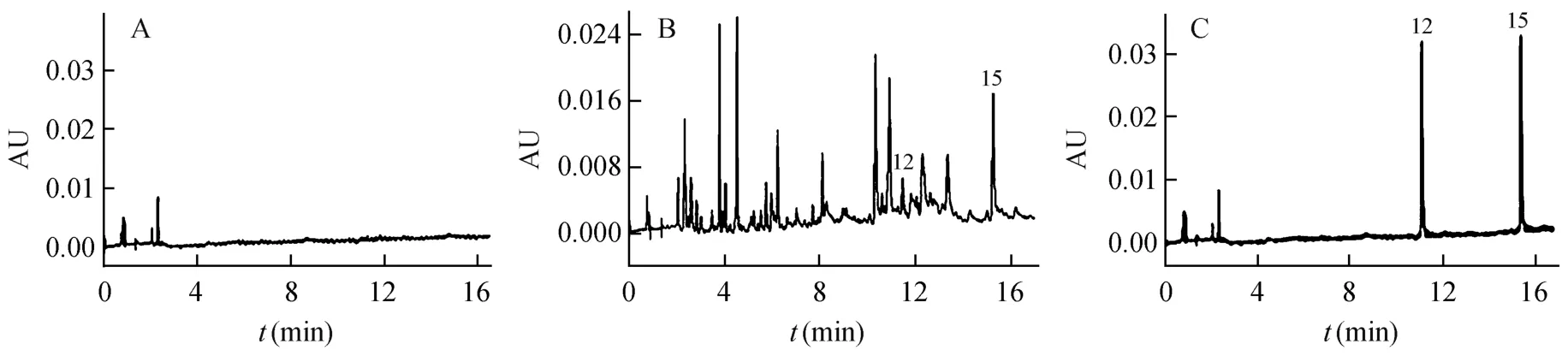
图1 UPC2色谱图(248 nm)A阴性;B补肾健脑颗粒(12 β-蜕皮甾酮;15松果菊苷);C对照品(12 β-蜕皮甾酮;15松果菊苷)Fig.1 Ultra performance convergence chromatograms(UPC2)(248 nm)of(A)negative;(B)Bushen Jiannao grains(12 β-ecdysterone;15 echinacoside)and(C)standard(12 β-ecdysterone standard;15 echinacoside)

表1 松果菊苷和β-蜕皮甾酮的线性关系Table 1 Linear relationship of echinacoside and β-ecdyserone
3.4.3 精密度、重复性、加样回收率试验分别对精密度、重复性、加样回收率进行考察,松果菊苷和β-蜕皮甾酮日内精密度的RSD分别为0.4%和0.3%;重复进样6次,两个化合物的峰面积RSD分别为0.5%和0.5%;加样回收实验(表2和表3)显示,松果菊苷和 β-蜕皮甾酮的加样回收率分别在99.7%~100.2%和99.8%~100.5%范围内。结果表明,本实验仪器精密度高,方法准确可靠、重复性好。
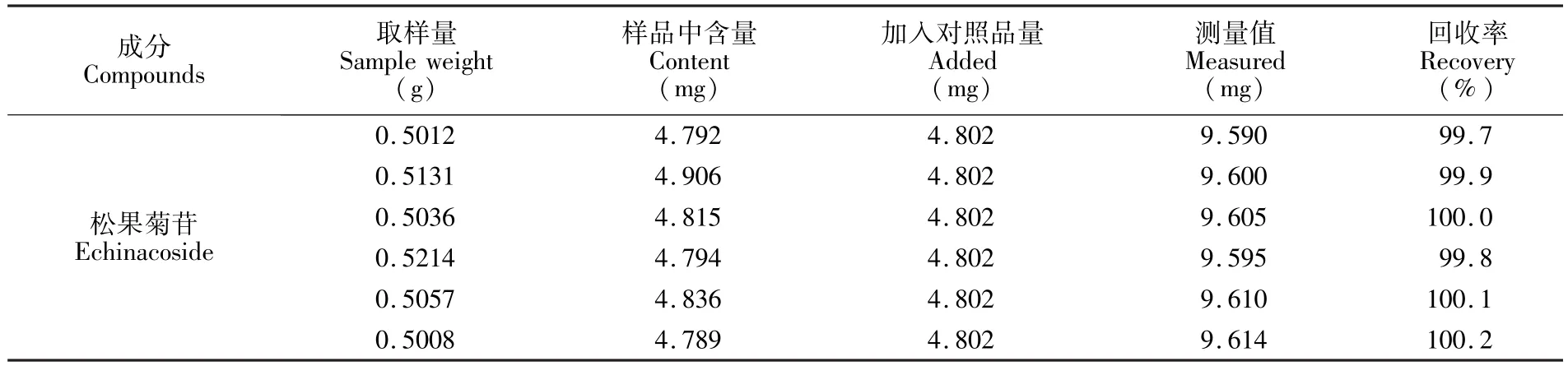
表2 松果菊苷加样回收实验结果Table 2 Recovery of echinacoside
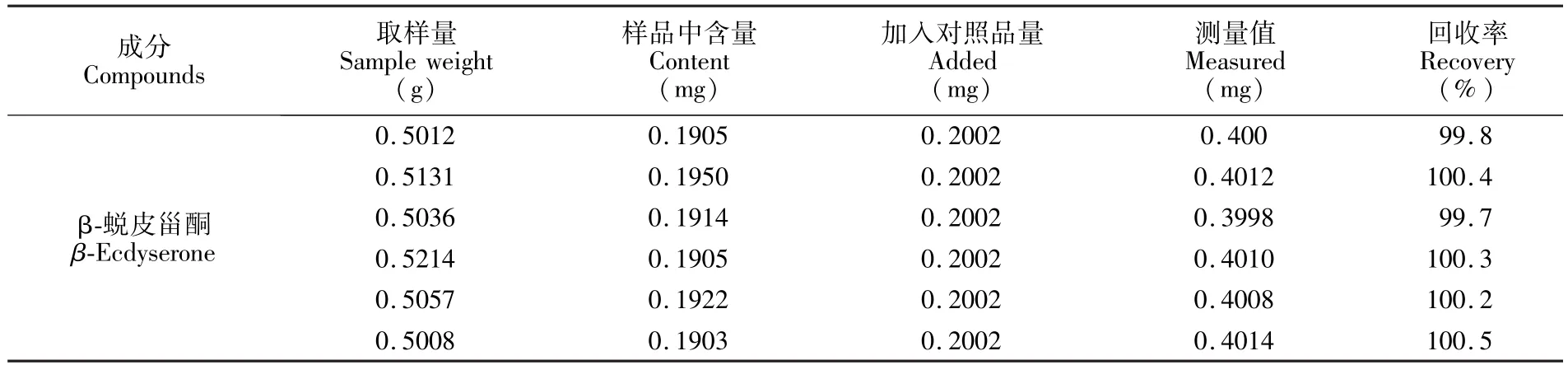
表3 β-蜕皮甾酮加样回收率实验结果Table 3 Recovery of β-ecdyserone
3.5 UPC2与HPLC、UPLC对比分析
取2.5节制备的补肾健脑颗粒供试品溶液,在优化条件下,分别用Waters高效液相色谱仪、超高效液相色谱仪和超高效合相色谱仪分析,248 nm下检测。HPLC和UPLC指纹图谱(图2A和2B)显示,两种方法都不能将补肾健脑颗粒成分有效分离,且峰很少。而UPC2法(图2C)与HPLC和UPLC法相比,样品出峰数量明显增多且各峰分离很好,松果菊苷和β-蜕皮甾酮峰均无干扰,且UPC2分析时间较HPLC缩短一半,充分体现了UPC2简便、快速、高分离度的特点。
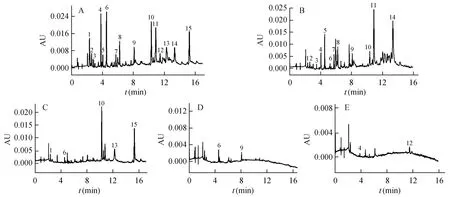
图2 UPC2色谱图(248 nm)(A)补肾键脑颗粒;(B)远志;(C)肉苁蓉;(D)石菖蒲;(E)怀牛膝Fig.2 Ultra performance convergence chromatogram(UPC2) (248 nm) of(A) Bushen Jiannao grains, (B)Polygala tenuifolia,(C)Cistanche and(D)Acorus graminus and(E)Achyranthes
3.6 样品的测定
取3批补肾健脑颗粒中试样品和各药材提取物适量,分别按2.4节制备供试品溶液,按2.2节方法检测,得到UPC2指纹图谱(图3)。计算补肾健脑颗粒中β-蜕皮甾酮和松果菊苷的含量,并通过保留时间和紫外光谱图归属其UPC2指纹图谱中主峰来源。结果表明,β-蜕皮甾酮(12号峰)平均含量为380 μg/g,松果菊苷(15号峰)平均含量为9.562 mg/g。各主峰来源明确,结果见表4。
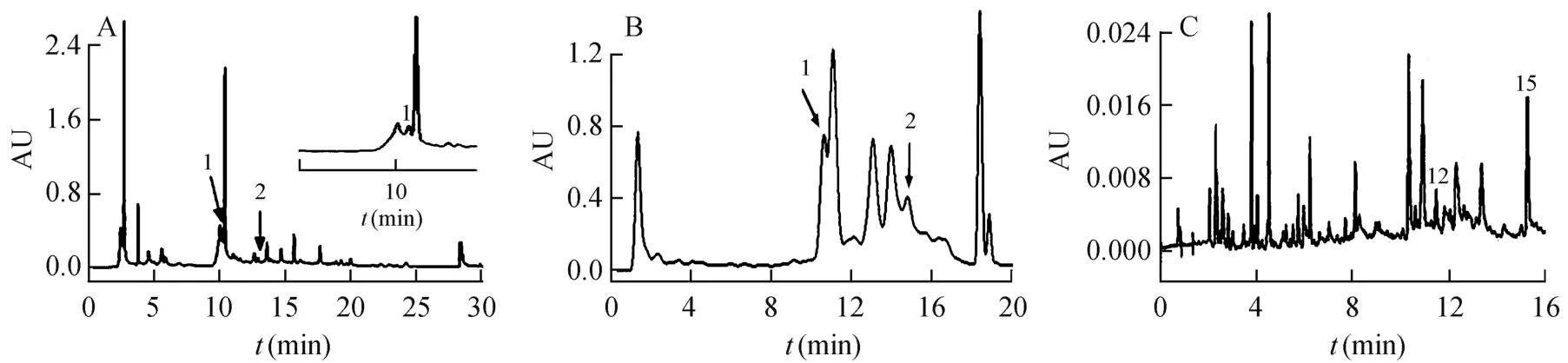
图3 色谱图(248 nm)A补肾健脑颗粒HPLC图(1,松果菊苷;2,β-蜕皮甾酮);B UPLC图(1,松果菊苷; 2,β-蜕皮甾酮);C UPC2图(12,β-蜕皮甾酮;15,松果菊苷)Fig.3 Chromatogram(248 nm).(A)HPLC chromatogram of bushen jiannao grains(1,echinacoside;2,β-ecdyserone);(B)UPLC chromatogram(1,echinacoside;2,β-ecdyserone);(C)UPC2chromatogram(12,β-ecdyserone; 15,echinacoside)
肉苁蓉的主要有效成分为苯乙醇苷类,其中松果菊苷含量较高[4~7],常以松果菊苷含量作为肉苁蓉质量控制指标。怀牛膝的主要活性成分为蜕皮甾酮等[8],常以β-蜕皮甾酮作为怀牛膝质量控制指标。石菖蒲的主要有效成分是α-细心醚和β-细辛醚[9],该制剂为水煎煮,无法检测到α-细心醚和β-细辛醚。远志的主要有效成分为皂苷类[9~11],文献[12~14]报道,远志和石菖蒲配伍时,远志中皂苷元含量大幅度降低,本实验尝试检测了远志皂苷,但在该制剂中未检测到。因此,本研究最终选择了以松果菊苷和β-蜕皮甾酮作为补肾健脑颗粒定量分析指标,指纹图谱中其他主要色谱峰作为定性分析指标。
本实验建立的UPC2方法,简便、快速,与HPLC和UPLC相比,既节省时间、溶剂,又能使补肾健脑颗粒样品峰完全分离,体现了其环保,高灵敏度,高分离度的优势。可用于复杂中药制剂质量的监测。
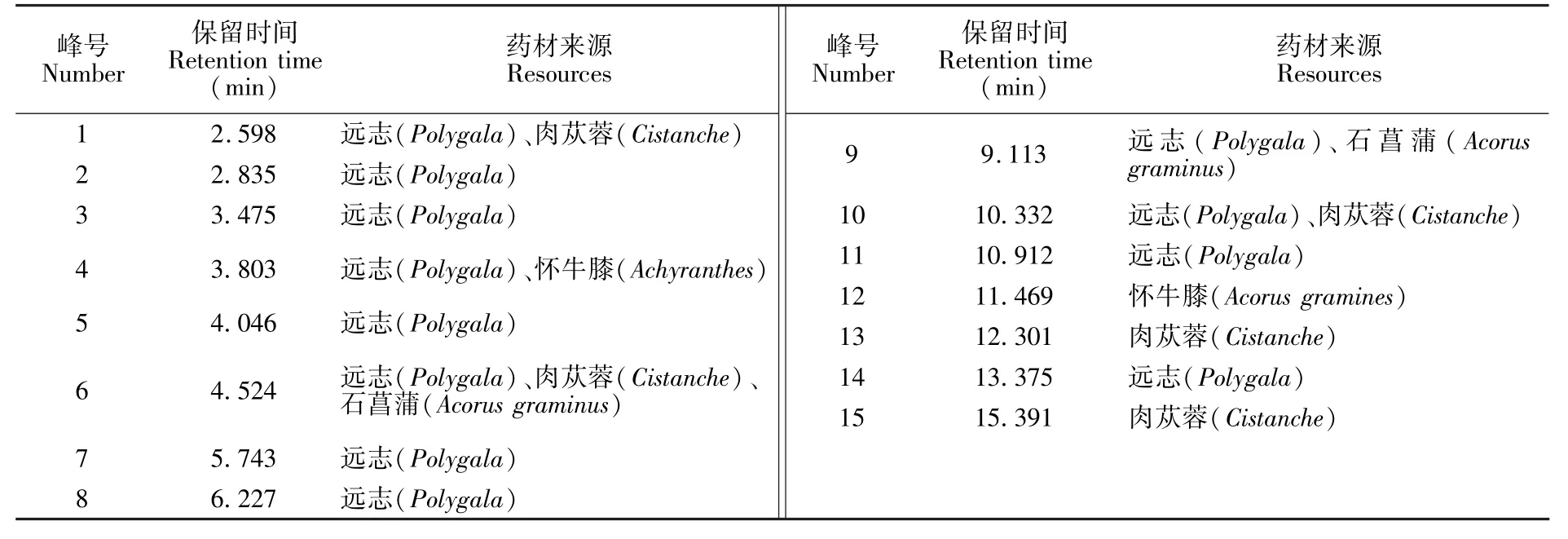
表4 各峰归属分析结果Table 4 Identification of each peak
1 SHI Jian-Gang.Journal of Pediatrics of Traditional Chinese Medicine.,2012,8(5):50-51
史建钢.中医儿科杂志,2012,8(5):50-51
2 XU Yong-Wei,SUN Qing-Long,HUNG Jing,TAN Xiao-Jie.Modern Instruments.,2012,18(5):45-48
徐永威,孙庆龙,黄静,谭晓杰.现代仪器,2012,18(5):45-48
3 LI Zhong-Hao,WU Shuai-Bin,LIU Shan-Shan,FAN Zi-Yan,YANG Fei,BIAN Zhao-Yang,TANG Gang-Ling,CHEN Zai-Gen,HU Qing-Yuan.Chinese J.Anal.Chem.,2013,41(12):1817-1824
李中皓,吴帅宾,刘珊珊,范子彦,杨 飞,边照阳,唐纲岭,陈再根,胡清源.分析化学,2013,41(12):1817-1824
4 LEI Li,SONG Zhi-Hong,TU Peng-Fei.Chinese Traditional and Herbal Drugs.,2003,34(5):473-476
雷丽,宋志宏,屠鹏飞.中草药,2003,34(5):473-476
5 LI Qing-Bao,YANG Lai-Xiu,YANG Shu-Qing.Inner Mongolia Medical Journal.,2003,35(6):537-538
李庆宝,杨来秀,杨树青.内蒙古医学杂志,2003,35(6):537-538
6 XU Chao-Hui,YANG Jun-Shan,LÜ Rui-Mian.Chinese Traditional and Herbal Drugs.,1999,30(4):244-246
许朝晖,杨峻山,吕瑞绵.中草药,1999,30(4):244-246
7 HU Jia-Qi,FENG Jia-Yuan.Clinical Journal of Chinese Medicine.,2012,15(4):26-28
胡佳琦,冯佳媛.中医临床研究,2012,15(4):26-28
8 ZHAO Wan-Ting,MENG Da-Li,LI Xian,LI Wei.Journal of Shenyang Pharmaceutical University.,2007,24(4): 207-210
赵婉婷,孟大利,李铣,李魏.沈阳药科大学学报,2007,24(4):207-210
9 JIANG Yong,TU Peng-Fei.Journal of Peking University(Health Sciences).,2004,36(1):94-98
姜勇,屠鹏飞.北京大学学报(医学版),2004,36(1):94-98
10 FU Jing,ZHANG Dong-Ming,CHEN Ruo-Yun.Chinese Traditional and Herbal Drugs.,2006,37(1):144-146
傅晶,张东明,陈若芸.中草药,2006,37(1):144-146
11 ZHANG Xiao-Ping.Heilongjiang Medicine Journal.,2004,17(2):139-140
张晓萍.黑龙江医药,2004,17(2):139-140
12 YANG Xiao-Yan,CHEN Fa-Kui.Journal of Shenyang Pharmaceutical University.,1999,16(1):71-78
杨晓燕,陈发奎.沈阳药科大学学报,1999,16,(1):71-78
13 ZHANG Wen-Juan,ZHENG Xiao-Hui,FANG Min-Feng,WANG Qing-Wei,CHEN Huan,LI Yun-Feng,ZHANG Yan. China Pharmaceuticals.,2009,18(15):6-7
张文娟,郑晓晖,房敏峰,王庆伟,陈焕,李云峰,张琰.中国药业,2009,18(15):6-7
14 HAN Yi-Li,LUO Feng-Juan,WANG Ying-Li.Chinese Journal of Information on Traditional Chinese Medicine.,2011, 18(3):67-68
韩毅丽,罗凤娟,王颖莉.中国中医药信息杂志,2011,18(3):67-68
药典委搭建开放、公益、合作实验平台新版药典亟需高精尖技术支持
近日,中国药典委员会在北京为其“ChP-Waters联合开放实验室”举行建设座谈会暨揭牌仪式。中国药典委员会秘书长张伟表示:“联合开放实验室的举办是中国药典委员会自身发展的需求,也是更好地开展药典标准制定、满足公众用药安全的需求,是中国药典委员会不断深化药品标准形成机制改革的新探索和新尝试。”
记者了解到,联合开放实验室作为一个公益项目,自宣布开建以来就获得行业的广泛关注。
张伟介绍,实验室聚集社会各种资源打造而成,旨在充分履行其开放、公益、创新和互利的使命,更好地服务和贡献于中国药典研究。“在实验室今后的发展运营中,仍然要坚持公益性,坚持为社会服务的宗旨,更希望有更多的企业和社会力量能加入到这一公益事业中来。”张伟强调。
“ChP-Waters联合开放实验室”设立于北京振东光明药物研究院实验大楼内,面积约400多平米。一方面将深入开展药典标准研究,检测方法和开发与验证工作;同时开展国内外药典标准的分析方法,及各论的数据比对工作。实验室还将为药品监管及药品生产科研人员提供培训,并就药物开发研究开展广泛的国际间技术交流。
鉴于实验室的开放性,各承担药典科研任务单位可根据需要,申请来人或带课题到实验室利用先进、新型的仪器设备进行试验研究。双方希望实验室最终成为中国药品标准领域的一个国家级技术支持中心。
沃特世(Waters®)公司和世界各地的药典委员会都有着非常广泛、深入的合作,各个国家的制药行业面临着不同的挑战。中国药典包括很多传统药物、中药,这些化合物的组成复杂得多,相应的分析和理解也就更困难。沃特世可以帮助中国药典针对这方面的内容,比如中药的分析、理解、验证、鉴别等,提供更高效的咨询服务、进行深度合作。”
Qualitative and Quantitative Analysis of Bushen Jiannao Grains by Ultra Performance Convergence Chromatography
ZHU Rui-Juan1,WANG Bo2,HU Fang1,LI Can1,LIU Xiao-Ya1,FENG Shi-Lan*1,ZHOU Wei*2
1(Department of Pharmacy,Lanzhou University,Lanzhou 730000,China)
2(Central Laboratory of Technical Gansu Entry-exit Inspection and Quarantine Bureall,Lanzhou 730000,China)
An ultra performance convergence chromatographic(UPC2)method was established for the attribution analysis of the main peaks as well as the quantitative determination of echinacoside and β-ecdyserone in Bushen Jiannao Grains.The samples were extracted with ethanol and separated on Waters ACQUITY UPC2TMBEH column(100 mm×3.00 mm,1.7 μm),with a gradient supercritical CO2-0.05% phosphoric acid-methanol solvent system at 40℃.The flow rate was 0.8 mL/min,the detection wavelength was set at 248 nm and the injection volume was 1 μL.Results showed that all the main peaks in the fingerprint were clearly attributed.The peak named 12 was β-ecdyserone with the content of 380 μg/g and the peak named 15 was echinacoside with the content of 9.562 mg/g.The method was simple,eco-friendly, accurate and reliable compared with HPLC and UPLC.
Ultra performance convergence chromatography;Bushen Jiannao Grains; Echinacoside; β-Ecdyserone
31 July 2014;accepted 18 September 2014)
2014-07-31收稿;2014-09-18接受
本文系兰州市科技三项经费项目(No.2012-2-70)资助
*E-mail:fengshl@lzu.edu.cn;zhouwei845@163.com
10.11895/j.issn.0253-3820.140655
