液相色谱法一次性检测多种甜味剂的测定研究
2015-05-28王婷婷
王婷婷

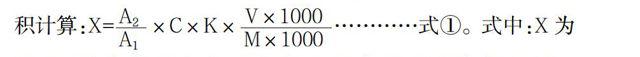
摘 要:本文建立了同时测定六种人工合成甜味剂阿斯巴甜、糖精钠、甜蜜素、安塞蜜、纽甜、甜菊糖甙的高效液相色谱分析方法,以Platicil ODS柱为分离柱,20mmol/L硫酸铵缓冲溶液(pH4.4)-乙腈为流动相,进行梯度洗脱,采用二极管阵列检测器进行检测,整个分离过程在30min内完成。样品加标回收率为85%~107%;相对标准偏差小于3.2%。该法可用于食品中六种甜味剂的同时测定。
关键词:高效液相色谱法;人工合成甜味剂;测定研究
一、前言
甜味剂是对能够赋予食品甜味的物质的总称。由于糖精钠(SA)、甜蜜素(SC)、安赛蜜(AK)、阿斯巴甜(ASP)、纽甜(NTM)、甜菊糖甙(S)等人工合成甜味剂具有高效、经济等优点,因此在食品中被广泛应用。但过量使用会造成一定的毒副作用。例如甜菊糖甙可能有一定的致癌作用;阿斯巴甜可影响苯丙酮尿病患者的发育;长期食用糖精钠会导致营养不良,其致癌可能性尚未完全排除;甜蜜素可能有致癌性,其代谢产物环己胺对心血管系统和睾丸有毒理作用。近年来发现有些商家对人工合成甜味剂的泛滥使用以及一些食品标签中未注明使用人工合成甜味剂,有些则以“糖蜜素”“甜味料”“蛋白糖”等混淆视听,以掩盖其产品的危害性,对消费者的身体健康构成危害,严重侵犯了广大消费者的利益。
二、实验部分
1.仪器与试剂
Agilent HP1100高效液相色谱仪(配有四元梯度泵、在线真空退气机、自动进样器、柱温箱、二极管阵列检测器DAD,美国安捷伦科技有限公司),AS3120A超声波振荡器(奥特塞恩斯仪器有限公司),pHS-3E型精密pH计(上海精密科学仪器有限公司),Platicil ODS色谱柱,SIGMA 3K15离心机,BP211D电子天平(Sartorius),ProElut PXC固相萃取柱。糖精钠(购置国家标准物质研究中心)、甜蜜素(Sigma公司产品)、安塞蜜、阿斯巴甜、甜菊糖甙、纽甜均为Supelco公司产品,乙腈(色谱纯),硫酸铵(优级纯);其余试剂均为分析纯;实验用水为超纯水系统制备。
2.色谱条件
色谱柱:ODS-C18(250mm×4.6mm,5μm);流动相为20mmol/L硫酸铵(用H3PO4调pH至4.4)-乙腈;梯度洗脱程序:乙腈:0~2min,5%;2~30min,5%~50%;流速0.8mL/min;柱温为30℃;检测波长为200nm,226nm;进样量为20μL。
3.样品前处理
所有樣品均为市场采集的样品,按照品种将其分为饮料、蜜饯、酱腌菜、酒类等,其中饮料按照国家标准进一步分为碳酸饮料、果汁、茶饮料、含乳饮料、植物蛋白饮料和固体饮料。
(1)介质简单的样品。例如碳酸饮料、果酒、葡萄酒等,称取10g样品(精确至0.001g),置于25mL容量瓶中,用水定容(如有乙醇需水浴加热除去乙醇后再用水定容至原体积)至刻度,混匀,经0.45μm滤膜过滤,滤液待上机测定。
(2)介质复杂的样品。如茶饮料、果汁等,称取6g样品(精确至0.001g),置于25mL容量瓶中,用水定容至刻度,4000r/min离心10min,取上清液,以下操作按2.4。
(3)介质为固体的样品。例如蜜饯、酱腌菜等,将样品粉碎,称取2g样品(精确至0.001g),称取适量置于25mL,加水后并超声振荡20min,加水定容至25mL。4000r/min离心10min,取上清液,以下操作按2.4。
(4)含乳饮料和植物蛋白饮料。称取6g样品(精确至0.001g),置于25mL容量瓶中,依次加入2mL0.08mol/LK4[Fe(CN)6](4.4)和2mL0.25mol/L乙酸锌(4.5)溶液,剧烈振荡2min,以沉淀蛋白质,加水定容至刻度,4000r/min离心10min,取上清液,4000r/min离心10min,取上清液,以下操作按2.4。
4.试样的净化
取上清液3mL经固相萃取柱(事先经3mL乙腈活化,再用3mL水平衡)萃取。然后依次用3mL20mmol/L硫酸铵和3mL乙腈洗脱,收集洗脱液于10mL容量瓶中,用水定容至刻度,混匀,经0.45μm滤膜过滤,滤液待上机测定。
5.测定
取处理液与混合标准使用液各20μL注入高效液相色谱分析仪进行分离,以保留时间定性,峰面积进行外标法定量。
6.计算公式
样品中糖精钠、甜蜜素、安赛蜜、阿斯巴甜、甜菊糖甙、纽甜含量(X)以毫克每千克(mg/kg)或毫克每升(mg/L)表示,按式①计算。其中糖精钠、甜蜜素、阿斯巴甜、甜菊糖甙、纽甜按200nm波长下的峰面积计算;安赛蜜按226nm波长下的峰面积计算:X=×C×K×…………式①。式中:X为样品中糖精钠、甜蜜素、安赛蜜、阿斯巴甜、甜菊糖甙、纽甜含量,单位为毫克每千克(mg/kg)或毫克每升(mg/L);A2为样品峰面积;A1为标液峰面积;K为稀释倍数;C为标液浓度,单位为微克每毫升(μg/mL);M为样品质量,单位为克(g);V为样品洗脱液浓缩定容体积,单位为毫升(mL)。
三、结果与讨论
1.检测波长的选择
本实验采用二极管阵列检测器,在190~400nm范围内对所有分析物进行光谱扫描。结果发现,六种甜味剂的最大吸收波长分别为:糖精钠204nm、甜蜜素194nm、安塞蜜226nm、阿斯巴甜202nm、甜聚糖甙200nm、纽甜206nm。选择200nm波长对糖精钠、甜蜜素、阿斯巴甜、甜聚糖甙、纽甜进行测定,选择226nm波长对安塞蜜进行测定。
2.流动相的选择
应为甲醇的截止波长为210nm左右,乙腈的截止波长为190nm左右,而糖精钠、甜蜜素、阿斯巴甜、甜聚糖甙、纽甜选择在200nm出检测。且乙腈的溶剂强度较高,粘度较小,故测定时选用乙腈。实验考察了乙酸胺、硫酸铵、柠檬酸铵等溶液作为缓冲溶液是对分离度的影响,结果表明,乙腈-硫酸铵缓冲体系的洗脱效果最好。当硫酸铵浓度分别为10mmol/L、20mmol/L、40mmol/L时,20mmol/L、40mmol/L分离效果较好,且二者相差不大,考虑到缓冲盐浓度增大,对色谱柱及系统管路造成的损害也会加大,所以选择20mmol/L的硫酸铵最为缓冲盐溶液。调节20mmol/L硫酸铵缓冲液的pH分别为3.0,4.0,4.4,5.0,6.0,在pH4.4条件下,六种甜味剂的色谱峰形和分离度良好。流动相配比对分离效果的影响实验结果表明,当硫酸铵缓冲液pH4.4,乙腈体积分数小于8%时,几种甜味剂均可实现基线分离,但阿斯巴甜和纽甜的保留时间比较长,峰形拖尾。随着乙腈体积分数的增大,所有分析物的保留时间均逐渐缩短。乙腈的体积分数大于17%时,安塞蜜、糖精钠和甜蜜素的色谱峰重叠,纽甜的保留时间大于40min。采用梯度洗脱可以缩短分析时间,提高分离度,改善峰形。考虑到本实验在短波长处测定,梯度增加太快会严重漂移,增加太慢分析时间就会加长,最终选择的梯度洗脱程序为乙腈:0~2min,5%;2~30min,5%~50%;流速0.8mL/min。在此色谱条件下,6种甜味剂的标准物质色谱图如图1。
图1 标准混合溶液的色谱图
3.线性范围、精密度和检出限
在实验条件下,所有分析物的质量浓度(mg/L)和峰面积(mAU)之间均有良好的线性关系,具体结果和检出限(S/N=3)见表1.对混合标准品(糖精钠、安塞蜜、阿斯巴甜、甜聚糖甙、纽甜均为40mg/L;甜蜜素80mg/L)平行测定6次,相对标准偏差见表1。
A:peak area response;C:concentration of analysis (mg/L).
4.实际样品的测定
(1)样品中甜味剂含量的测定:在该实验条件下,分别对5种样品进行了测定,结果见表2。
表2 实际样品的测定结果(n=3)
(2)样品加标回收率的测定。取适量样品,在线性范围内加入一定量的标准品做加标回收实验。每种样品平行测定3次,测定结果见表3。平均加标回收率在85%~107%,相对标准偏差小于3.2%,测定结果令人满意。
四、小结
本文通过对多种甜味剂高效液相色谱法的实验分析,提出了适用于食品中多种甜味剂的检测方法,以消除对商品中单一甜味剂检测而造成其他甜味剂漏检的可能性,同时减少同种食品多种甜味剂检测时前处理的重复性,更加省时、快捷,也为质检、海关等部门净化国内外市场,保障食品安全提供更好的科学依据。
参考文献:
[1]白艳玲,王丽玲.超声提取气相色谱法快速测定食品中甜蜜素含量的研究[J].中国热带医学,2004,4(2):190-191.
[2]陈金东,李蔚高效液相色谱法同时测定食品中阿斯巴甜、阿力甜[J].中国卫生检验杂志,2006(9):225-227.
