活性炭固相萃取/胶束电动色谱联用技术用于雷公藤3种有效成分的测定
2015-04-27蒋银燕郭丽娟崔小莹王翠琼胡小建李建明
蒋银燕,郭丽娟,崔小莹,王翠琼,胡小建,李建明
(1.长沙医学院 基础医学院,湖南 长沙 410219;2.中南大学 湘雅医学院,湖南 长沙 410013)
活性炭固相萃取/胶束电动色谱联用技术用于雷公藤3种有效成分的测定
蒋银燕1,郭丽娟1,崔小莹1,王翠琼1,胡小建1,李建明2*
(1.长沙医学院 基础医学院,湖南 长沙 410219;2.中南大学 湘雅医学院,湖南 长沙 410013)
建立了活性炭固相萃取/胶束电动色谱法同时测定中药雷公藤中的雷公藤甲素、雷公藤内酯酮和雷酚内酯的方法。优化了萃取pH值、乙醇洗脱体积、电泳运行缓冲溶液pH值与浓度、胶束SDS的浓度及进样时间、电压等条件。进行电泳分离之前,分析物用活性炭进行吸附后用2.0 mL乙醇洗脱。在214 nm 波长处,分离电压20 kV,20 mmol/L硼酸-10 mmol/L硼砂(pH 8.0)-20 mmol/L SDS运行缓冲溶液条件下,3种成分在8 min内得到完全分离,雷公藤甲素、雷公藤内酯酮在4.0×10-5~4.0×10-3mol/L,雷酚内酯在4.0×10-5~1.0×10-3mol/L浓度范围内线性关系良好,其回收率为81.0%~102.9%,方法的检出限分别为1.42×10-6,7.90×10-7,2.96×10-7mol/L,相对标准偏差(RSD )分别为 3.2%,5.4%,3.5%。所建立的方法简单、快速、准确,成功用于雷公藤片样品的测定。
雷公藤甲素;雷公藤内酯酮;雷酚内酯;固相萃取;胶束电动色谱
雷公藤(Tripterygium wilfordii Hook F.)系卫矛科雷公藤属木制藤本植物,作为杀虫剂使用已有近2 000年的历史。近年发现该药有抗炎和抑制免疫的功效,目前已用于人体免疫系统疾病的治疗,包括:类风湿关节炎、强直性脊髓炎、儿童原发性和继发性肾病综合症、成人各型肾炎、系统性红斑狼疮、血管炎和银屑病等多种皮肤病[1-3]。雷公藤及其提取物的成分很复杂,早在1972年,Kupchan就试图分离提取雷公藤中的有效成分。国内外学者认为雷公藤红素(Tripterine)、雷公藤内酯酮(Triptonide)、雷公藤甲素(又名雷公藤内酯醇,Triptolide)和雷公藤乙素(Tripdiolide)是雷公藤的主要活性成分[4]。
目前测定中药中有效成分的常用方法有气相色谱法(GC)[5-6]、固定化脂质体色谱(ILC)[7]、高效液相色谱法(HPLC)[8-9]及毛细管电泳法(CE)[10-11]等。毛细管电泳作为一种新兴的高效分析技术,在中药鉴定分析方面以其高效快速、样品用量少、操作简便等优点得到广泛使用,但由于毛细管进样体积小(进样量小于毛细管长度的1%)以及柱上检测的光程短,通用型的紫外检测器虽然能达到很低的质量检出限,但样品的浓度检出限仍很高(比HPLC高几个数量级)[12-13],这使CE在实际样品痕量组分的分离分析中受到很大限制。因此,在使用CE进行定量分析测定时需对样品富集后再检测,以提高其检测灵敏度,同时消除基体干扰。据报道,与毛细管电泳法联用的样品预富集方法主要有液液萃取(LLE)[14]、固相萃取(SPE)[15-16]以及浊点萃取[17]等。但尚未见采用活性炭萃取富集后用胶束电动色谱技术来分离测定雷公藤内酯酮、雷公藤甲素及雷酚内酯的报道。本文利用胶束电动色谱的高效、快速、运行成本低等特点,联用活性炭固相萃取技术,建立了一种能够同时检测雷公藤中上述3种有效活性成分(结构式见图1)的分析方法。
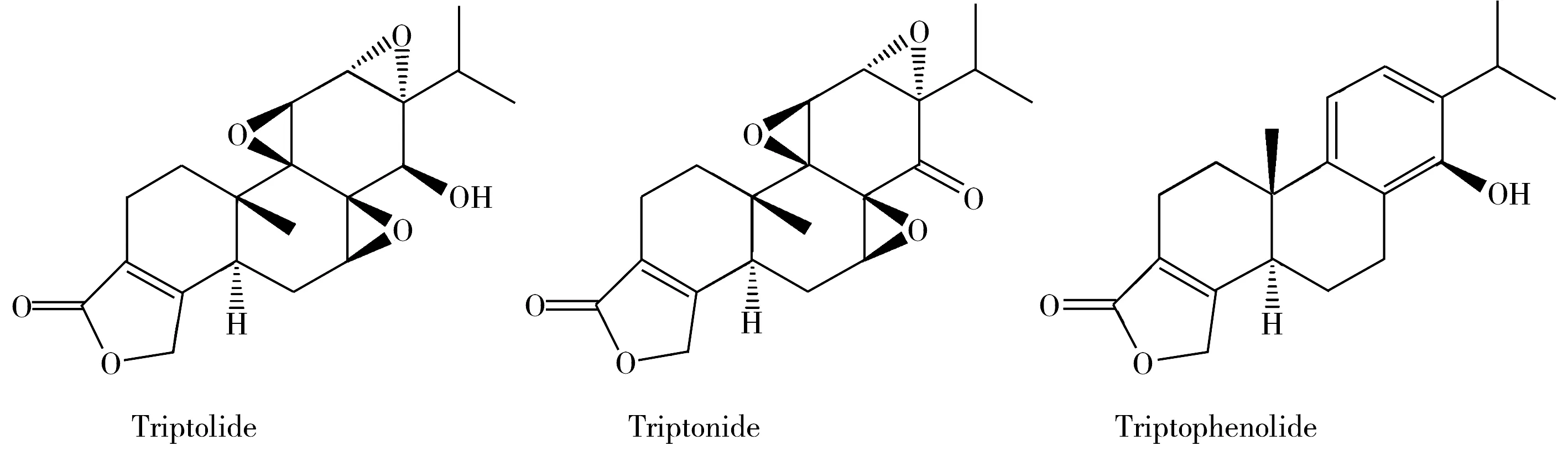
图1 3种雷公藤有效成分的化学结构式
1 实验部分
1.1 仪器与试剂
高效毛细管电泳仪带紫外检测器(北京彩陆科学仪器有限公司);未涂敷熔融石英毛细管(75 μm I.D.×375 μm O.D.,柱长51 cm,有效长度43 cm,河北永年科技有限公司)。
雷公藤内酯酮、雷公藤甲素、雷酚内酯(色谱纯,福建医药厂),雷公藤片(三金制药厂,湖北黄石,雷公藤甲素标识含量为33 μg/片)。实验所用其他试剂均为分析纯,实验用水为二次蒸馏水。所有溶液均使用0.22 μm纤维膜过滤且超声3 min。
1.2 溶液的配制
1.2.1 标准溶液的配制 分别精密称取一定量的雷公藤内酯酮、雷公藤甲素和雷酚内酯标准品,溶于甲醇中,配成浓度为4.0×10-3mol/L的储备液。实验时,工作溶液由储备液用甲醇稀释至所需浓度。
1.2.2 样品溶液的配制 精确称取雷公藤片一片置于离心管中,用1 mL甲醇浸泡24 h,再将混合物超声30 min后,以3 500 r·min-1离心20 min。上层液体用0.22 μm的微孔滤膜过滤,所得清液用乙醇定容至2.0 mL。
1.3 实验方法
1.3.1 活性炭固相萃取 在10 mL离心管中加入10 mL Na2HPO4-KH2PO4缓冲溶液(pH 6.5),再移取一定量样品溶液至离心管中,加入30 mg活性炭后随即超声15 min,再以3 500 r/min离心5 min,弃去上层液体,固相中加入2 mL乙醇,超声洗脱后,离心5 min,用注射器将管中液体吸取出来,经0.22 μm微孔滤膜过滤至小试剂瓶中待用。
1.3.2 MEKC分离条件 实验前,毛细管依次用1 mol/L NaOH、无水乙醇、水、运行缓冲液冲洗2 min,以保证实验的重现性。实验所用溶液进样前均用0.22 μm的滤膜过滤,并经超声波脱气约3 min。运行缓冲溶液:20 mmol/L硼酸-10 mmol/L硼砂-20 mmol/L SDS缓冲溶液(pH 8.0),检测波长214 nm,分离电压20 kV,高度进样,进样高度约9 cm,进样时间5 s,为确保重复性,每个样品至少平行测定3次。

图2 pH值对萃取率的影响

图3 缓冲溶液pH值对电泳迁移时间的影响
2 结果与讨论
2.1 活性炭固相萃取条件的优化
2.1.1 pH值的影响 萃取体系的pH值是影响固相萃取效率的重要因素,pH值可以改变目标物/吸附剂的离子化或质子化程度。为了考察不同pH值的磷酸缓冲液对萃取率的影响,本实验研究了pH值在4.0~8.0范围内变化时对萃取率的影响情况,结果如图2所示。从图中可见,pH 6.5时3种化合物的萃取率均较高,萃取率可达90%以上。
2.1.2 超声洗脱时间的影响 超声洗脱时间对萃取率有一定的影响。实验发现,萃取率随时间的增加而增加,但当洗脱时间超过15 min后,3种有效成分的萃取效率随时间增加出现缓慢降低现象。综合考虑,本实验选择最佳超声洗脱时间为15 min。
2.1.3 乙醇洗脱体积的影响 考察了乙醇洗脱体积对萃取率的影响,分别加入1.0,1.5,2.0,2.5,3.0 mL乙醇后超声15 min,以3 500 r/min离心5 min。取出离心管,吸取出溶液,通过0.22 μm的微孔滤膜过滤后,进行CE测定。结果显示,最初萃取率随着洗脱溶液体积的增加而增大,当用2.0 mL乙醇洗脱时,3类成分的萃取效率均可达到90%以上,继续增大洗脱溶液体积,则萃取效率反而有所降低。因此,本实验选择乙醇的最佳洗脱体积为2.0 mL。
2.2 MEKC条件的优化
2.2.1 缓冲溶液pH值的影响 缓冲溶液的pH值直接影响毛细管的表面电势,从而影响电渗流的速度,同时溶液的pH值也决定样品中各组分分子的解离程度,从而影响组分的迁移时间和分离度。实验发现pH值在7.0~10.0范围内能实现3种成分的有效分离,在该范围内,分离时间随pH值的变化结果如图3所示。当pH值小于8.0时,由于电渗流随着pH值的增加而增大,因此各分析物的出峰时间也随之缩短。缓冲液的pH值大于8.0后,各分析物的出峰时间反而延长。因此,实验选用pH值为8.0的缓冲体系。
2.2.2 缓冲溶液种类及浓度的影响 缓冲溶液种类及浓度对胶束电动色谱分离效果有明显的影响。在pH 8.0条件下,研究了不同种类的缓冲溶液对分离效果的影响。实验发现,硼砂-硼酸缓冲体系中3种成分的分离效果明显,灵敏度较高。另一方面,缓冲溶液浓度决定了溶液的粘度、扩散系数及毛细管内壁的电渗流,是影响分离度的重要因素。对不同浓度的缓冲溶液(硼酸浓度:10~40 mmol/L,硼砂浓度:5~25 mmol/L)进行考察后发现,分离度和迁移时间均随缓冲液浓度的增加而增加,且高浓度的缓冲液会使毛细管内壁产生过高的焦耳热,从而使管内产生气泡,或者使基线不稳定,甚至干扰分离。3种成分在20 mmol/L硼酸-10 mmol/L硼砂缓冲液中运行时可得到较为理想的峰形。

图4 SDS浓度对迁移时间的影响
2.2.3 SDS浓度对分离的影响 考察了SDS浓度在5~50 mmol/L变化时对电泳分离的影响。如图4所示,由于胶束的交联作用,分离度以及分离时间均随SDS浓度的增加而增加,但当SDS浓度低于10 mmol/L时,溶剂和内酯醇的峰无法分离,且雷酚内酯不出峰;当SDS浓度低于20 mmol/L时,出现双峰。综合考虑分离度和分析时间,本实验选用SDS的最佳浓度为20 mmol/L。
2.2.4 进样时间及分离电压的影响 考察了进样时间在3~10 s范围内变化时对峰形以及峰面积的影响。实验结果表明,在3~5 s内,随着进样时间的增长,进样量增大,峰面积呈线性增大,但分离度变化不大;而在5~10 s内,进样时间越长,进样量越大,峰面积越大,但分离度减小,色谱峰展宽。为了获得最佳分离效果,最终选择5 s作为理想的进样时间。
溶质迁移时间、柱效和分离度均可从升高外加电压而获益。实验结果表明:随着电压的增大,分离时间呈线性减小,但当电压增至20 kV时,迁移时间开始偏离线性,峰形重合,分离度减小。这是由于在高电压下,产生的焦耳热增多,在不能有效地驱散所产生的焦耳热的情况下,柱温显著升高,导致缓冲溶液的电导增加,电流增大,粘度减小,双电层增厚,且毛细管内径形成径向温度梯度,导致区带增宽,所以本实验选择最佳分离电压为20 kV。
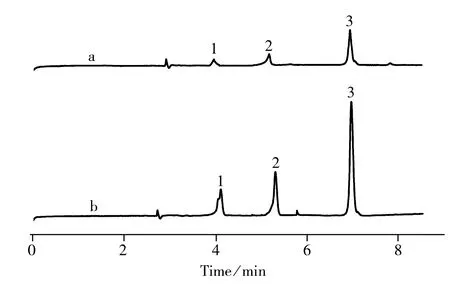
图5 3种雷公藤成分的标样电泳图
2.3 线性范围、检出限与精密度
分别用SPE/MEKC联用方法和MEKC法测定3种相同浓度的雷公藤成分,电泳图如图5所示。比较所得的电泳图可以看出,相同浓度的3种雷公藤成分经固相萃取后,峰面积均有明显增加,富集倍数为5倍,说明固相萃取法具有显著的富集效果。在优化实验条件下,3种成分在8 min内得到完全分离。将浓度均为5.0×10-4mol/L的3种成分的混合标准溶液连续进样3次,峰面积的相对标准偏差(RSD)为2.5%~5.5%,结果表明此方法稳定、重现性良好。以标准溶液浓度对相应峰面积绘制标准曲线,得到3种化合物的线性范围、回归方程、相关系数及检出限(3倍信噪比,S/N=3)见表1。从表1中可以看出,雷公藤甲素、雷公藤内酯酮在4.0×10-5~4.0×10-3mol/L范围内,雷酚内酯在4.0×10-5~1.0×10-3mol/L范围内线性关系良好,方法的检出限为2.96×10-7~1.42×10-6mol/L,峰面积的相对标准偏差(RSD,n=6 )均小于5.5%,方法显示了良好的精密度。
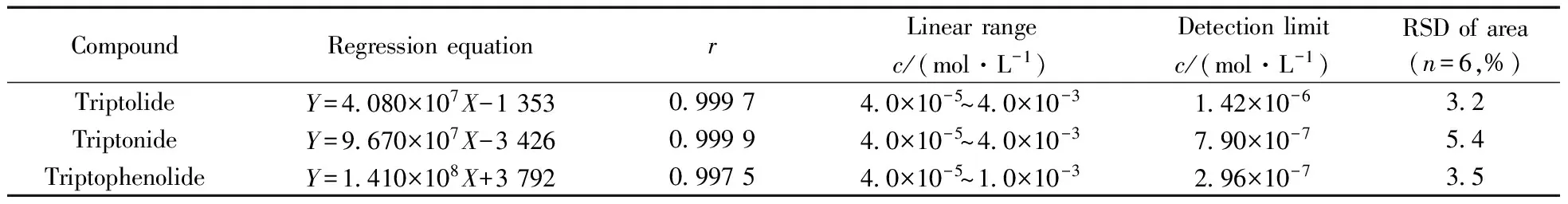
表1 3种有效成分的线性范围、回归方程、相关系数及检出限

图6 雷公藤片样品(a)及加标样品(b)的电泳图
2.4 实际样品的测定
雷公藤片样品溶液经“1.2.2”方法处理后,用所建立的方法进行其3种有效成分含量的测定,并进行加标回收实验。结果显示,在优化的分离和检测条件下,基本能够消除雷公藤片中基体杂质对分离检测的干扰,雷公藤片样品溶液中所含雷公藤甲素为3.95×10-5mol·L-1(即28.5 μg/片,与标示值33 μg/片接近),不含雷公藤内酯酮及雷酚内酯成分(图6)。加标测定后,3种雷公藤成分的加标回收率为81.0%~102.9%(见表2),基本满足分析方法的要求。

表2 实际样品的测定结果及加标回收率
-:no data
3 结 论
本文采用活性炭固相萃取/胶束电动色谱联用技术,建立了中药雷公藤中雷公藤甲素、雷公藤内酯酮和雷酚内酯的分离测定方法,检出限为2.96×10-7~1.42×10-6mol/L。用活性炭作为固相萃取介质,建立的SPE/MEKC联用方法操作简单,且进样量小、分析时间短、试剂消耗少、环保,为中药雷公藤提取物的分离检测提供了快速、高效、准确的分析方法。将该方法用于雷公藤片样品的测定,结果较为满意。
[1] Ma W G,Zhang T,Zhang C,Shang J H,Zhu L D.ChinaJ.Tradit.Chin.Med.Pharm.(马伟光,张滔,张超,尚建华,祝丽娣.中华中医药杂志),2006,21(2):117-120.
[2] Brinker A M,Raskin I.J.Chromatogr.A,2005,1070:65-70.
[3] Li B Z,Bai J,Yang G L,Li Z W,Wang L J,Chen Y.J.Chromatogr.A,2005,1097:199-202.
[4] Liu C A,Peng M.AnticancerChineseHerbDictionary.Wuhan:Science and Technology Publishing Company(刘春安,彭明.抗癌中草药大辞典.武汉:科学技术出版社),1994.
[5] Liu Q,Kong W,Qiu F,Wei J,Yang S,Zheng Y,Yang M.J.Chromatogr.B,2012,885/886(15):90-96.
[6] Xu R,Wu J,Liu Y,Zhao R,Chen B,Yang M,Chen J.Chemosphere,2011,84(7):908-912.
[7] Wang Y,Kong L,Lei X,Hu L,Zou H,Welbeck E,Bligh S W A,Wang Z.J.Chromatogr.A,2009,1216(11):2185-2191.
[8] Xiao S,Luo K,Wen X,Fan X,Cheng Y.J.Pharm.Biomed.,2014,92(15):82-89.
[9] Yan Y,Hao Y,Hu S,Chen X,Bai X.J.Chromatogr.A,2013,1322:8-17.
[10] Zhu Q,Xu X,Huang Y,Xu L,Chen G.J.Chromatogr.A,2012,1246:35-39.
[11] Zhao H,Chen Z.J.Chromatogr.A,2014,1340:139-145.
[12] Cerutti S,Silva M F,Gásquez J A,Olsina R A,Martínez L D.Electrophoresis,2005,26(18):3500-3506.
[13] Wu Y W,Jiang Y Y,Liu J F,Xiong K.Electrophoresis,2008,29:819-826.
[14] Sung I H,Lee Y W,Chung D S.Anal.Chim.Acta,2014,838:45-50.
[15] Mai T D,Bomastyk B,Duong H A,Pham H V,Hauser P C.Anal.Chim.Acta,2012,727:1-7.
[16] Chen D D,He D X,Qi K Z,Lu C Z.J.Instrum.Anal.(陈玎玎,何东旭,祁克宗,陆翠珍.分析测试学报),2012,31(10):1334-1338.
[17] Zhong S,Tan S N,Ge L,Wang W,Chen J.Talanta,2011,85(1):488-492.
Determination of Three Active Components in Tripterygium Wilfordii Hook F.by Activated Carbon Solid Phase Extraction Combined with Micellar Electrokinetic Capillary Chromatography
JIANG Yin-yan1,GUO Li-juan1,CUI Xiao-ying1,WANG Cui-qiong1,HU Xiao-jian1,LI Jian-ming2*
(1.Department of Basic Medicine,Changsha Medicial University,Changsha 410219,China;2.Xiangya School of Medicine,Central South University,Changsha 410013,China)
A method has been developed for simultaneous determination of triptolide,triptonide and triptophenolide in traditional Chinese herb Tripterygium wilfordii Hook F.by micellar electrokinetic capillary chromatography(MEKC) combined with activated carbon solid phase extraction.Effects of some important factors,such as pH value of extraction,eluant volume of ethanol,pH value and concentration of running buffer,concentration of sodium dodecyl sulfate(SDS),separation voltage and injection time were investigated.The analytes were extracted with activated carbon in phosphate buffer solution(pH 6.5),then eluted with 2.0 mL ethanol before separated by MEKC.At 214 nm wavelength,the detection analytes could be well separated within 8 min at separation voltage of 20 kV in 20 mmol/L boric acid-10 mmol/L borax(pH 8.0)-20 mmol/L SDS.Under the optimized conditions,the calibration curves were linear in the range of 4.0×10-5-4.0×10-3mol/L for triptolide and the triptonide,and 4.0×10-5-1.0×10-3mol/L for triptophenolide.The detection limits of triptolide,triptonide and triptophenolide were 1.42×10-6,7.90×10-7,2.96×10-7mol/L,respectively,and the relative standard deviations(RSDs,n=6) for peak areas were 3.2%,5.4%,3.5%,respectively.The proposed method had the advatages of simplicity,rapidness,low sample consumption and accuracy,and was successfully applied in the determination of the analytes in Tripterygium wilfordii tablet with recoveroes of 81.0%-102.9%.
triptolide;triptonide;triptophenolide;solid phase extraction(SPE);micellar electro-kinetic capillary chromatography(MEKC)
2014-09-30;
2014-10-25
湖南省科技厅科技计划项目(2014SK3025);湖南省教育厅科学研究项目(12C0489,12B018,13C1125 );长沙市科技计划项目(K1207042-31)
10.3969/j.issn.1004-4957.2015.02.011
O657.7;TQ460.72
A
1004-4957(2015)02-0189-05
*通讯作者:李建明,在读博士,副教授,研究方向:神经生物学,Tel:0731-88498021,E-mail:jyyhello@126.com
