桥头碳上氮杂芳基功能化的双吡唑甲烷与羰基钨反应
2015-04-01丁可孙遵明李厚谦唐良富
丁可 孙遵明 李厚谦 唐良富
(南开大学化学系,元素有机化学国家重点实验室,天津300071)
桥头碳上氮杂芳基功能化的双吡唑甲烷与羰基钨反应
丁可 孙遵明 李厚谦 唐良富*
(南开大学化学系,元素有机化学国家重点实验室,天津300071)
研究了(氮甲基咪唑-2-基)双(3,5-二甲基吡唑)甲烷(L1),2-吡啶基双(3,5-二甲基吡唑)甲烷(L2)及4-吡啶基双(3,5-二甲基吡唑)甲烷(L3)与羰基钨的反应,合成了一系列以单齿,双齿及三齿氮配位的羰基金属衍生物LW(CO)5(L=L1或L3),LW(CO)4(L=L1,L2或L3)和LW(CO)3(L=L1或L2)。核磁,红外及X-射线单晶衍射分析表明这3种配体表现出了可变的配位方式。在LW(CO)5中,当配体为L1时,其倾向于通过咪唑氮与金属配位,而为L3则倾向于利用吡啶氮与金属作用;在LW(CO)4中,配体L1表现为通过咪唑氮和吡唑氮原子配位的[N,N′]双齿配体,而L2和L3表现为通过吡唑氮原子配位的[N,N]双齿配体;在LW(CO)3中,L1和L2起着[N,N,N′]三齿螯合配体的作用。
氮配体;双吡唑甲烷;咪唑;吡啶;钨
As one of the most popular nitrogen-containing donor ligands,bis(pyrazol-1-yl)methane has been used extensively in bioinorganic,coordination chemistry and organometallic fields[1].The modification of bis (pyrazol-1-yl)methane on the bridgehead carbon has drawn extensive attention in recent years,because thegenerated heteroscorpionate ligands show good donor properties and variable coordination behavior towards main group and transition metals[2-4].For example, azaaryl functionalized bis(pyrazol-1-yl)methane on the methinecarbonhasbeenexploredtoprepare interesting metal supramolecular frameworks[5-8]as well as highly efficient catalysts or catalyst precursors[9-13]. Owing to the existence of the extra coordinate site, these azaaryl functionalized bis(pyrazol-1-yl)methanes can simultaneously supply hard donors(such as pyrazolyl nitrogen)and relatively soft donors(such as pyridyl nitrogen),which is further advantageous to adjust the coordination environments around the metal centers.Ourpreviousinvestigationsshowedthat functional bis(pyrazol-1-yl)methanes on the methine carbon atom displayed unusualreactivity[14-17].For example,thereactionofarylfunctionalizedbis (pyrazol-1-yl)methanes with W(CO)5THF resulted in the cleavage of a Csp3-N bond to form novel pyrazole derivatives[16].Reaction of bis(3,5-dimethylpyrazol-1-yl) methylthiolate with Fe3(CO)12gave rise to the formation of unexpected(3,5-dimethylpyrazol-1-yl)dithioformate derivatives[15].As part of our ongoing interest in functional bis(pyrazol-1-yl)methanes,herein we report the reaction of azaaryl functionalized bis(pyrazol-1-yl) methanes with tungsten carbonyl,which yielded a series of tungsten derivatives with mono-,bi-and tridentate ligands depending on different reaction conditions.
1 Experimental
All reactions were carried out under an atmosphere of argon.Solvents were dried and distilled prior to use according to standard procedures.NMR(1H and13C)were recorded on a Bruker 400 spectrometer using CDCl3as solvent unless otherwise noted,and the chemical shifts were reported with respect to the reference(internal SiMe4for1H and13C NMR).IR spectra were obtained as KBr pellets on a Nicolet 380 spectrometer.Elemental analyses were carried out on an Elementar Vairo EL analyzer.(N-Methylimidazol-2 -yl)bis(3,5-dimethylpyrazol-1-yl)methane(L1)[10-11],(pyridin-2-yl)bis(3,5-dimethylpyrazol-1-yl)methane(L2)[10-11]andbis(3,5-dimethylpyrazol-1-yl)methanone[18]were prepared by the published methods.
1.1 Synthesis of(pyridin-4-yl)bis(3,5-dimethylpyrazol-1-yl)methane(L3)
Themixtureofbis(3,5-dimethylpyrazol-1-yl) methanone(2.20 g,10 mmol),isonicotinaldehyde (0.95 mL,10 mmol)and CoCl2·6H2O(10 mg,0.042 mmol)was stirred and heated at 50℃for 2 h.After cooling to room temperature,CH2Cl2(40 mL)was added to the reaction mixture.The CH2Cl2solution was washed with water three times(3×30 mL),and dried over anhydrous MgSO4.After removing of the solvent,the residue was recrystallized from CH2Cl2/ hexane to give white crystals of L3.Yield:1.3 g (46%).1H NMR:δ 2.21(s,6H),2.22(s,6H)(CH3), 5.88(s,2H,H4of pyrazole),6.88(d,J=5.4 Hz,2H), 8.60(d,J=6.0 Hz,2H)(C5H4N),7.50(s,1H,CH). Anal.Calcd.for C16H19N5(%):C 68.30,H 6.81,N 24.89;Found(%):C 68.62,H 6.68,N 25.16.
1.2 Reaction of L1with W(CO)6
A solution of W(CO)6(0.18 g,0.5 mmol)and L1(0.14 g,0.5 mmol)dissolved in THF(25 mL)was irradiated with a 300 W high-pressure Hg lamp for 10 h at room temperature.The solvent was removed in vacuo,and the residue was isolated by column chromatography on silica with ethyl acetate/hexane(1∶1,V/V)as the eluent firstly to give complex 1.Then ethyl acetate was used as the eluent to give complex 2.Recrystallization of complexes 1 and 2 from CH2Cl2/ hexane afforded yellow crystals.
Data for 1,yield:30 mg(10%).1H NMR:δ 2.04 (s,6H),2.15(s,6H)(CH3),3.31(s,3H,NCH3),5.93 (s,2H,H4of pyrazole),6.86(d,J=1.4 Hz,1H),7.27 (d,J=1.4 Hz,1H)(protons of imidazole),7.80(s,1H, CH).13C NMR:δ 10.4,13.9(CH3),35.1(NCH3),69.6 (CH),107.7(C4of pyrazole),124.3,134.0,134.5, 145.0,149.0(carbons of imidazole as well as C3and C5of pyrazole),197.9(4C),201.6(1C)(CO).IR ν(C≡O):2 071(m),1 976(sh),1 909(vs,br)cm-1.Anal. Calcd.for C20H20N6O5W(%):C 39.49,H 3.31,N 13.82; Found(%):C 39.31,H 3.25,N 13.84.
Data for 2,yield:0.10 g(35%).1H NMR:δ 1.48 (s,3H),2.08(s,3H),2.48(s,3H),2.63(s,3H)(CH3),3.40(s,3H,NCH3),5.84(s,1H),6.17(s,1H)(H4of pyrazole),6.97(s,1H),7.16(s,1H)(protons of imidazole),7.40(s,1H,CH).13C NMR:δ 10.2,12.4,13.6, 17.8(CH3),34.0(NCH3),63.8(CH),109.2,109.7(C4of pyrazole),122.5,133.3,140.1,140.2,144.1,150.1, 156.9(carbons of imidazole as well as C3and C5of pyrazole),202.5,203.3,212.9,213.1(CO).IR ν(C≡O):2005(s),1 877(vs),1 840(vs),1 789(vs)cm-1. Anal.Calcd.for C19H20N6O4W·CH2Cl2(%):C 36.11,H 3.33,N 12.63;Found(%):C 35.82,H 3.31,N 12.44.
1.3 Reaction of L1with W(CO)5THF
L1(0.14 g,0.5 mmol)was added to the solution of W(CO)5THF in THF,prepared in situ by irradiation of a solution of W(CO)6(0.18 g,0.5 mmol)in THF(25 mL)for 8 h.The resulting reaction mixture was stirred and heated at reflux for 24 h.After cooling to room temperature,the yellow solids were filtered,and washed with acetone to give complex 3.Yield:0.15 g (54%).1H NMR(DMSO-d6):δ 2.43(s,6H),2.55(s, 6H)(CH3),3.94(s,3H,NCH3),6.16(s,2H,H4of pyrazole),7.19(d,J=1.2 Hz,1H),7.31(d,J=1.2 Hz, 1H)(protons of imidazole),7.32(s,1H,CH).13C NMR (DMSO-d6):δ 10.7,15.4(CH3),33.5(NCH3),57.9(CH), 106.9(C4of pyrazole),123.3,130.6,138.9,141.5,153.1 (carbons of imidazole as well as C3and C5of pyrazole), 223.3(1C),224.0(2C)(CO).IR ν(C≡O):1 895(s), 1 749(vs,br)cm-1.Anal.Calcd.for C18H20N6O3W(%): C 39.15,H 3.65,N 15.22;Found(%):C 39.15,H 3.37,N 15.41.
Complex 3 was also obtained by heating 1 or 2 at reflux in THF in good yields(87%for 1 and 92%for 2,respectively).
1.4 Reaction of L2with W(CO)6
This reaction was carried out as above-mentioned reaction of L1with W(CO)6,but L1was replaced by L2. Complex 4 was obtained in 25%yield.1H NMR:δ 2.41(s,6H),2.51(s,6H)(CH3),6.10(s,2H,H4of pyrazole),6.28(d,J=7.9 Hz,1H),7.21~7.23(m,1H), 7.61(td,J=1.7 and 7.9 Hz,1H),8.51(d,J=7.9 Hz, 1H)(protons of pyridyl),7.19(s,1H,CH).13C NMR:δ 11.8,17.3(CH3),69.1(CH),108.1(C4of pyrazole), 120.5,124.6,138.0,142.8,150.7,155.8,162.3(carbons of pyridyl as well as C3and C5of pyrazole),202.2, 203.5,212.0(2C)(CO).IRν(C≡O):2004(s),1878(vs), 1 856(vs),1 813(vs)cm-1.Anal.Calcd.for C20H19N5O4W (%):C 41.61,H 3.32,N 12.13;Found(%):C 41.86,H 3.48,N 12.54.
1.5 Heating 4 in refluxing THF
Complex 4(50 mg,0.087 mmol)was dissolved in THF(15 mL).The resulting solution was heated at reflux for 8 h.After cooling to room temperature,the red solids were filtered,and washed with CH2Cl2to give complex 5.Yield:40 mg(87%).1H NMR (DMSO-d6):δ 2.42(s,6H),2.58(s,6H)(CH3),6.21(s, 2H,H4of pyrazole),7.50(t,J=6.9 Hz,1H),8.13(t,J= 6.9 Hz,1H),8.47(d,J=7.8 Hz,1H),9.01(d,J=7.5 Hz,1H)(protons of pyridyl),7.74(s,1H,CH).13C NMR (DMSO-d6):δ 10.8,15.3(CH3),66.7(CH),107.3(C4of pyrazole),125.6,125.9,139.9,141.9,151.2,153.0, 155.5(carbons of pyridyl as well as C3and C5of pyrazole),202.7,211.8(2C)(CO).IR ν(C≡O):1 889 (vs),1 758(vs,br)cm-1.Anal.Calcd.for C19H19N5O3W· 0.5CH2Cl2(%):C 39.58,H 3.41,N 11.84;Found(%): C 39.22,H 3.38,N 12.09.
1.6 Reaction of L3with W(CO)6
This reaction was carried out as above-mentioned reaction of L1with W(CO)6,but L1was replaced by L3. When the mixture of ethyl acetate/hexane(2∶3,V/V) was firstly used as theeluent,complex6was obtained.Then acetone was used as the eluent to give complex 7.
Data for 6,yield:22%.1H NMR:δ 2.21(s,12H, CH3),5.92(s,2H,H4of pyrazole),6.85(d,J=5.6 Hz, 2H),8.78(d,J=5.6 Hz,2H)(protons of pyridyl),7.48 (s,1H,CH).13C NMR:δ 11.5,13.6(CH3),72.1(CH), 107.7(C4of pyrazole),124.1,141.0,147.6,149.3,156.0 (carbons of pyridyl as well as C3and C5of pyrazole), 198.7(4C),202.3(1C)(CO).IR ν(C≡O):2 072(m), 1 980(s),1 926(sh),1 903(vs)cm-1.Anal.Calcd.for C21H19N5O5W(%):C 41.67,H 3.16,N 11.57;Found (%):C 41.50,H 3.32,N 11.58.
Data for 7,yield:48%.1H NMR(DMSO-d6):δ 2.08(s,6H),2.17(s,6H)(CH3),5.97(s,2H,H4of pyrazole),7.07(d,J=5.7 Hz,2H),8.98(d,J=5.7 Hz, 2H)(protons of pyridyl),7.86(s,1H,CH).13C NMR (DMSO-d6):δ 10.9,13.4(CH3),69.9(CH),105.6(C4of pyrazole),124.8,140.6,147.3,147.7,156.2(carbons of pyridyl as well as C3and C5of pyrazole),198.4(CO). IR ν(C≡O):2 002(m),1 872(vs),1 847(vs),1 804 (vs)cm-1.Anal.Calcd.for C20H19N5O4W(%):C 41.61, H 3.32,N 12.13;Found(%):C 41.34,H 3.21,N 11.79.
1.7 Reaction of L3with W(CO)5THF
This reaction was carried out as above-mentioned reaction of L1with W(CO)5THF,but L1was replaced by L3.This reaction afforded complexes 6 in 19% yield and 7 in 34%yield,respectively.
1.8 X-ray crystallography
Yellow crystals of 2 and red crystals of 4 and 7 suitable for X-ray analysis were grown by slow diffusion of hexane into their CH2Cl2solutions at-18℃.All intensity data were collected with a SCX-MINI CCD diffractometer for 2 and 7 as well as Rigaku Saturn CCD diffractometer for 4 using graphite monochromated Mo Kα radiation(λ=0.071 073 nm).Semiempirical absorption corrections were applied using the Crystalclear program[19].The structures were solved by direct methods and difference Fourier map using SHELXS of the SHELXTL package and refined with SHELXL[20]by full-matrix least-squares on F2.Crystals of 2 and 4 contained one CH2Cl2molecule.Crystals of 7 contained one water molecule.Furthermore,the hydrogen atoms of the water ligand in 7 could not be properly determined.All nonhydrogen atoms were refined anisotropically.Hydrogen atoms were added geometrically and refined with riding model position parameters.A summary of the fundamental crystal data for these complexes is listed in Table 1.
CCDC:1030402,2;1030403,4;1030404,7.
2 Results and discussion
2.1 Reaction of(N-methylimidazol-2-yl)bis(3,5-dimethylpyrazol-1-yl)methane
(N-Methylimidazol-2-yl)bis(3,5-dimethylpyrazol-1-yl)methane(L1)has been used as a chelating tridentate ligand in previous works[11].Herein,we find that this ligand can act as a monodentate ligand through the imidazolyl nitrogen or a chelating bidentate ligand by the imidazolyl nitrogen and one pyrazolyl nitrogen atom.Reaction of L1with W(CO)6under the irradiation of high-pressure Hg lamp gave complexes 1 and 2 (Scheme 1),according to the results of their NMR and IR spectroscopy.In addition,these two complexes could be transformed to complex 3 when heated in refluxing THF.Complex 3 was also obtained by the direct reaction of L1with W(CO)5THF under refluxing conditions.

Table 1Crystallographic data and refinement parameters for complexes 2,4 and 7
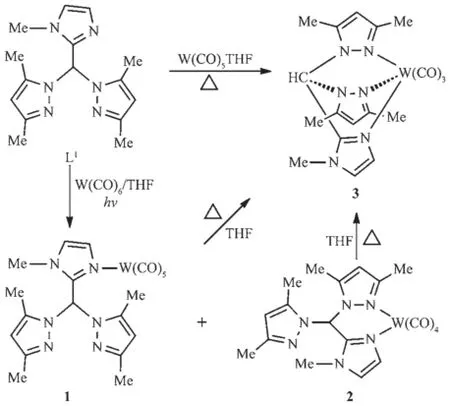
Scheme 1Reaction of(N-methylimidazol-2-yl)bis(3,5-dimethylpyrazol-1-yl)methane
Complexes 1~3 have been characterized by IR and NMR spectroscopy.They showed significantly different IR and NMR spectra.A ν(C≡O)band at 2 071 cm-1corresponding to the A1eqmode for the pseudo C4vmetal center in the M(CO)5fragment[21]was observed in the IR spectrum of 1,implying that L1acted as a monodentate ligand in this complex.While complex 2 displayed four carbonyl absorption peaks in its IR spectrum,consistent with a typical cis-tetracarbonyl arrangement[22].In addition,the IR spectrum of 3 matched those of the tricarbonyltungsten species.The NMR spectra of 1~3 also supportthe suggested structures.The13C NMR spectrum of 1 revealed two carbonyl carbon signals with ca.a 1∶4 intensity ratio, correspondingtoamonosubstitutedpentacarbonyl skeleton.Furthermore,its1H and13C NMR spectra disclosed two equivalent pyrazolyl rings,indicating that the donor atom was the imidazolyl nitrogen instead of the pyrazolyl nitrogen.In contrast,the13C NMR spectrum of 2 revealed four carbonyl carbon signalswiththesameintensity.Meanwhile,two inequivalent pyrazolyl rings were observed in its1H and13C NMR spectra.These results suggested that L1acted as a chelating bidentate ligand in 2 through the imidazolyl nitrogen and one pyrazolyl nitrogen.A chelating tridentate coordination mode of L1can be used to give explanation of the NMR spectra of 3.For example,two carbonyl carbon signals with ca.a 1∶2 intensity ratio and two equivalent pyrazolyl signals were observed in its13C or1H NMR spectrum owing to the tridentate coordination mode of L1.
The structure of 2 has been further confirmed by X-ray structural analyses,and is presented in Fig.1. The selected bond distances and angles are listed in Table 2.As above-mentioned by1H NMR,L1coordinates to the tungsten atom through the imidazolyl nitrogen andonepyrazolylnitrogenatom,givingasixmembered metallacycle with a boat conformation.The uncoordinated pyrazolylgroupoccupiestheaxial position of the boat conformation,possibly avoiding the steric repulsion to the methyl groups of coordinated imidazolylandpyrazolylrings.Twocis-carbonyl groups are distorted with the angles W(1)-C(1)-O(1)of 170.7(6)°and W(1)-C(3)-O(3)of 170.5(8)°.The angleC(1)-W(1)-C(3)of 169.8(3)°markedly deviates from linearity.These data reflect the presence of the large steric repulsion between these carbonyls with the ligand. The W-Nimidazolylbond distance is 0.224 2(5)nm,slightly shorterthanthe W-Npyrazolylbond distance(0.2303(5)nm), butcomparable to those reported in other carbonyl tungsten derivatives with imidazolyl ligands[23-24].

Fig.1 Molecular structure of 2 with 30%probability displacement ellipsoids

Table 2 Selected bond distances(nm)and angles(°)for complexes 2,4 and 7
2.2 Reaction of pyridylbis(3,5-dimethylpyrazol-1-yl)methanes
Irradiation of the solution of 2-pyridyl functionalized bis(pyrazol-1-yl)methane(L2)and W(CO)6in THF using high-pressure Hg lamp gave only complex 4, which could be transformed to complex 5 when heated in refluxing THF(Scheme 2).Similar treatment of 4-pyridyl analogs(L3)and W(CO)6yielded complexes 6 and 7.While 6 could not be converted to 7 even heated at high temperature.All these four complexes were fully characterized by IR and NMR spectroscopy, and the structures of 4 and 7 were unambiguously confirmed by X-ray crystallography.

Scheme 2Reaction of pyridylbis(3,5-dimethylpyrazol-1-yl)methanes
The IR spectra of 4 and 7 were similar to that of 2.They displayed analogous carbonyl carbon signals in their13C NMR spectra.Like 3,complex 5 showed twostrongcarbonylabsorptionbandsinitsIR spectrum,and two carbonyl carbon signals in its13C NMR spectrum.As anticipated,a νCOband assigned to the A1eqmode for the pseudo C4vmetal center was observed in the IR spectrum of 6.The corresponding carbonyl absorption occurred at 2 072 cm-1,very close to the value in 1.Moreover,complexes 1 and 6 showed very analogous chemical shifts of carbonyl carbon atoms in their13C NMR spectra.These results suggest that complexes 4,7 and 2,5 and 3,6 and 1 should possess similar carbonyl metal fragments.It is worth pointing out that a proton of the pyridyl group significantly shifted upfield(δ 6.28)in 4,compared with that of the free L2(δ 6.92)[12].A similar result was observed in 2,in which one methyl proton signal appeared in the high field(δ 1.48).The upfield shift of these protons should be attributed to the shielding effect of one pyrazolyl ring,as disclosed by the X-ray crystal structural analyses of 2 and 4.Fig.1 reveals thatthemethylprotonsat5-postionofthe uncoordinated pyrazolyl ring locate over the plane of the coordinated pyrazolyl group.Fig.2 shows that the proton at 3-postion of the pyridyl group is situated over the N(2)-N(1)-C(6)-C(7)-C(8)pyrazolyl plane. This shielding phenomenon has found in other aryl functionalized bis(pyrazol-1-yl)methane derivatives[16].

Fig.2Molecular structure of 4 with 30%probability displacement ellipsoids
The molecular structures of 4 and 7 are shown in Figs.2 and 3,respectively.These two complexes possess a similar fundamental framework.For example,L2and L3coordinate to the tungsten atom in a bidentate chelating fashion,and the uncoordinated pyridyl group occupies the axial position of the boat conformation, like the uncoordinated pyrazolyl group in 2.The crystal data reveal that complexes 4 and 7 share some analogous structural parameters,such as similar WNpyrazolylbond distances and N-W-N bite angles(see Table 2).These W-Npyrazolyl(0.224 7~0.227 4 nm)bond distances in 4 and 7 are comparable to those reported forcarbonyltungstenderivativesbearingN,N-bidentate chelating bis(pyrazol-1-yl)methane ligands, such as average 0.224 2 nm in(2-MeOC6H4)CH(pz)2W (CO)4and 0.227 1 nm in(2-MeOC6H4)CH(3,5-Me2Pz)2W(CO)4(pz=pyrazol-1-yl)[16].Two cis-carbonyls in these two complexes deviate significantly from linearity. Moreover,the angle of W(1)-C(4)-O(4)in 7(167.9(5)°) issmallerthanthecorrespondinganglesin4 (169.1(3)°)and 2(170.7(6)°).Additionally,the angle of C(2)-W(1)-C(4)in 4(165.4(2)°)is slightly smaller than the corresponding angle in 7(166.8(7)°),and smallerthantheangleofC(1)-W(1)-C(3)in 2(169.8(3)°). These data reflect the presence of the larger steric repulsion between the ligands with two cis-carbonyls in 4 and 7 compared with 2.

Fig.3Molecular structure of 7 with 30%probability displacement ellipsoids
In summary,we have investigated the reaction of azaaryl functionalized bis(pyrazol-1-yl)methanes withW(CO)6,which gave rise to the formation of a series of new tungsten complexes with mono-,bi-and tridentate ligands.The different donor ability of imidazolyl,pyridyl and pyrazolyl nitrogen atoms as well as the controllable reaction conditions possibly plays important roles for the structural diversity of these derivatives.
[1]Pettinari C,Pettinari R.Coord.Chem.Rev.,2005,249:663-691
[2]Otero A,Fernández-Baeza J,Lara-Sánchez A,et al.Coord. Chem.Rev.,2013,257:1806-1868
[3]Otero A,Fernández-Baeza J,Lara-Sánchez A,et al.Eur.J. Inorg.Chem.,2008:5309-5326
[4]Higgs T C,Carrano C J.Inorg.Chem.,1997,36:291-297
[5]Dura G,Carrion,M C,Jalon,F A,et al.Cryst.Growth Des., 2014,14:3510-3529
[6]Xiao C H,Liu J C,Song X Y,et al.Transition Met.Chem., 2013,38:307-311
[7]Carrion,M C,Dura G,Jalon,F A,et al.Cryst.Growth Des., 2012,12:1952-1969
[8]Reger D L,Gardinier J R,Grattan T C,et al.J.Organomet. Chem.,2005,690:1901-1912
[9]Hoffmann A,Flrke U,Schürmann M,et al.Eur.J.Org.Chem., 2010:4136-4144
[10]Zhang J,Li A,Hor T S A.Dalton Trans.,2009:9327-9333
[11]Zhang J,Li A,Hor T S A.Organometallics,2009,28:2935-2937
[12]Arroyo N,la Torre F G,Jalón F A,et al.J.Organomet. Chem.,2000,603:174-184
[13]Byers P K,Canty A J,Honeyman R T.J.Organomet.Chem., 1990,385:417-427
[14]Sun J P,Zhao D W,Song H B,et al.Organometallics, 2014,33:4425-4432
[15]Liu X L,Zhang X Y,Song H B,et al.Organometallics, 2012,31:5108-5113
[16]Ding K,Cheng C H,Yang Y X,et al.J.Organomet.Chem., 2011,696:3662-3667
[17]ZHANG Xiao-Yan(张晓燕),SONG Hai-Bin(宋海斌),TANG Liang-Fu(唐良富).Acta Chim.Sinica(化学学报),2011,69: 2567-2573
[18]Carrión M C,Jalón F A,Manzano B R,et al.Eur.J.Inorg. Chem.,2007:3961-3973
[19]CrystalStructure3.7.0andCrystalclear1.36:Crystal Structure Analysis Package,Rigaku and Rigaku/MSC(2000) TX.
[20]Sheldrick G M.Acta Crystallogr.,2008,A64:112-114
[21]Kraihanzel C S,Cotton F A.Inorg.Chem.,1963,2:533-540
[22]Orgel L E.Inorg.Chem.,1962,1:25-29
[23]ZHANG Xiao-Yan(张晓燕),DING Ke(丁可),SONG Hai-Bin (宋海斌),et al.Chinese J.Inorg.Chem.(无机化学学报), 2010,26:1-7
[24]Li H J,Liu X L,Ding K,et al.J.Organomet.Chem.,2014, 757:8-13
Reaction of Tungsten Carbonyl with Bis(pyrazol-1-yl)methanes Functionalized by Azaaryl Groups
DING KeSUN Zun-MingLI Hou-QianTANG Liang-Fu*
(Department of Chemistry,State Key Laboratory of Elemento-Organic Chemistry,Nankai University,Tianjin 300071,China)
Reaction of W(CO)6with(N-methylimidazol-2-yl)bis(3,5-dimethylpyrazol-1-yl)methane(L1),(pyridin-2-yl)bis(3,5-dimethylpyrazol-1-yl)methane(L2)and(pyridin-4-yl)bis(3,5-dimethylpyrazol-1-yl)methane(L3)yielded complexes LW(CO)5(L=L1or L3),LW(CO)4(L=L1,L2or L3)and LW(CO)3(L=L1or L2),respectively.NMR,IR and X-ray structural analyses indicated that these three ligands possessed variable coordination modes in these complexes.L1and L3acted as monodentate ligands through the imidazolyl nitrogen or the pyridyl nitrogen in LW (CO)5.A N,N′-chelating bidentate ligand through the imidazolyl nitrogen and one pyrazolyl nitrogen was observed in L1W(CO)4,while L2and L3acted as N,N-chelating bidentate ligands through two pyrazolyl nitrogens in L2W (CO)4and L3W(CO)4.A tridentate N,N,N′-chelating ligand through two pyrazolyl nitrogens and the imidazolyl or 2-pyridyl nitrogen was observed in L1W(CO)3and L2W(CO)3.The different donor ability of these imidazolyl, pyridyl and pyrazolyl nitrogens possibly plays important roles for the structural diversity.CCDC:1030402,2; 1030403,4;1030404,7.
N ligand;bis(pyrazol-1-yl)methane;imidazole;pyridine;tungsten
O614.61+3
A
1001-4861(2015)02-0345-08
10.11862/CJIC.2015.058
2014-11-04。收修改稿日期:2014-11-28。
国家自然科学基金(No.21372124)和NFFTBS(No.J1103306)资助项目。
*通讯联系人。E-mail:lftang@nankai.edu.cn;会员登记号:S060015703M。
