R基团搜索技术用于2-氨基-6-磺酰苯甲腈衍生物的分子设计
2015-02-22仝建波
仝建波, 白 敏, 赵 翔, 常 佳
(1.陕西科技大学 化学与化工学院, 陕西 西安 710021; 2.陕西科技大学 教育部轻化工助剂化学与技术重点实验室, 陕西 西安 710021)
R基团搜索技术用于2-氨基-6-磺酰苯甲腈衍生物的分子设计
仝建波1,2, 白敏1, 赵翔1, 常佳1
(1.陕西科技大学 化学与化工学院, 陕西 西安710021; 2.陕西科技大学 教育部轻化工助剂化学与技术重点实验室, 陕西 西安710021)
摘要:艾滋病对人类的健康构成了很大的威胁,因此,抗艾滋病药物的研究设计成为了当今社会的重要任务之一.本文采用基于R基团搜索技术的Topomer CoMFA 方法,对60个2-氨基-6-磺酰苯甲腈衍生物分子进行了三维定量构效关系分析.所得优化模型的拟合、交互验证及外部验证的复相关系数分别为0.851、0.601和0.671;并采用Topomer Search技术对ZINC数据库进行R基团的虚拟筛选,最终得到5个Ra基团和2个Rb 基团,并设计出了10个新化合物.研究结果表明:所建立的Topomer CoMFA模型具有良好的稳定性和预测能力;基于R基团的Topomer Search技术可以有效地筛选并设计出新的化合物,这为抗艾滋病新药设计提供了依据.
关键词:Topomer CoMFA; 2-氨基-6-磺酰苯甲腈衍生物; 定量构效关系; Topomer Search; 新药设计
0引言
艾滋病(Acquired Immunodeficiency Syndrome,AIDS)是由人类免疫缺陷病毒(Human Immunodeficiency Virus,HIV)感染而引起的全身性传染病[1].HIV是一种 RNA 逆转录病毒,根据血清学反应与病毒核酸序列测定,HIV 可分为Ⅰ型(HIV-1)和 Ⅱ型(HIV-2)[2,3],两者之间有着一定的免疫交叉反应.艾滋病绝大多数由 HIV-1 引起.HIV-1 的感染过程主要包括以下步骤:病毒粒子对宿主细胞的依附、辅受体相互作用、病毒与细胞的结合、病毒RNA的逆转录、前病毒DNA的整合、DNA的转录、病毒蛋白质的表达、病毒的组装以及病毒粒子的发芽与成熟[4-6]等.以上几个过程中所涉及到的酶是抗艾滋病药物研发的靶点.
目前,美国FDA批准用于HIV-1感染者临床治疗的药物共有31种,主要包括核苷类逆转录酶抑制剂、非核苷类逆转录酶抑制剂、蛋白酶抑制剂、整合酶抑制剂和进入抑制剂等.但由于HIV的易突变性导致很多药物不断地失效,因此寻找并研究新的、有效的抗艾滋病药物受到了社会各界的广泛关注.
本研究采用Topomer CoMFA[7,8]方法对一系列2-氨基-6-磺酰苯甲腈衍生物进行了定量构效关系的研究(QSAR)[9],这可为抗艾滋病药物的设计奠定一定基础.
Topomer CoMFA方法是第二代CoMFA[10]方法,此方法将目标分子切割成两个或多个结构片段,计算每个Topomer片段的立体场和静电场性质,再采用偏最小二乘(PLS)[11]回归分析方法对每个片段分别建立CoMFA模型.所建立的Topomer CoMFA模型结合 Topomer Search[12,13]技术,可以进行基于配体的虚拟筛选,从而为新药的设计奠定基础.
不同于CoMFA的地方是,Topomer CoMFA通过一系列完全客观一致的叠合规则完成 3D-QSAR 分析的准备工作;此外,Topomer CoMFA与CoMFA相比,还具有重复性高的优势.利用Topomer CoMFA方法,可以快速建立预测模型并进行分析与评价,这为同类小分子抑制剂的结构优化提供了理论依据[14].
1方法与步骤
1.1 数据集选择与划分
60个2-氨基-6-磺酰苯甲腈衍生物的分子结构与活性数据见表 1[15-17]所示.活性标度为pIC50(-logIC50),其中IC50值为HIV-1病毒感染细胞半数抑制浓度.按照随机化原则,将数据集划分为训练集(42个化合物)和测试集(18个化合物).其中,训练集用来建立模型,测试集用来验证模型的外部预测能力.
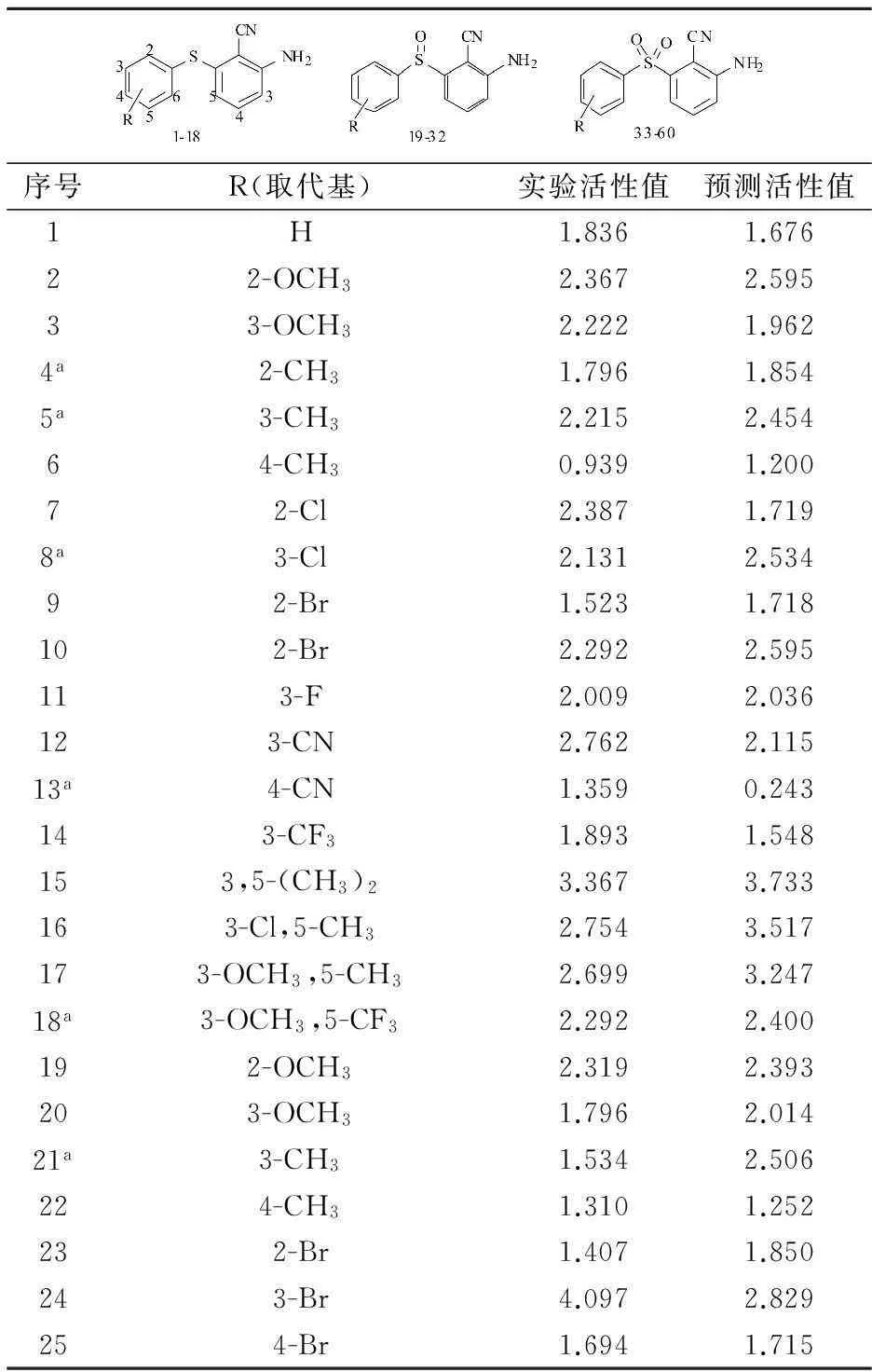
表1 60个2-氨基-6-磺酰苯甲腈衍生物的分子结构及其活性值
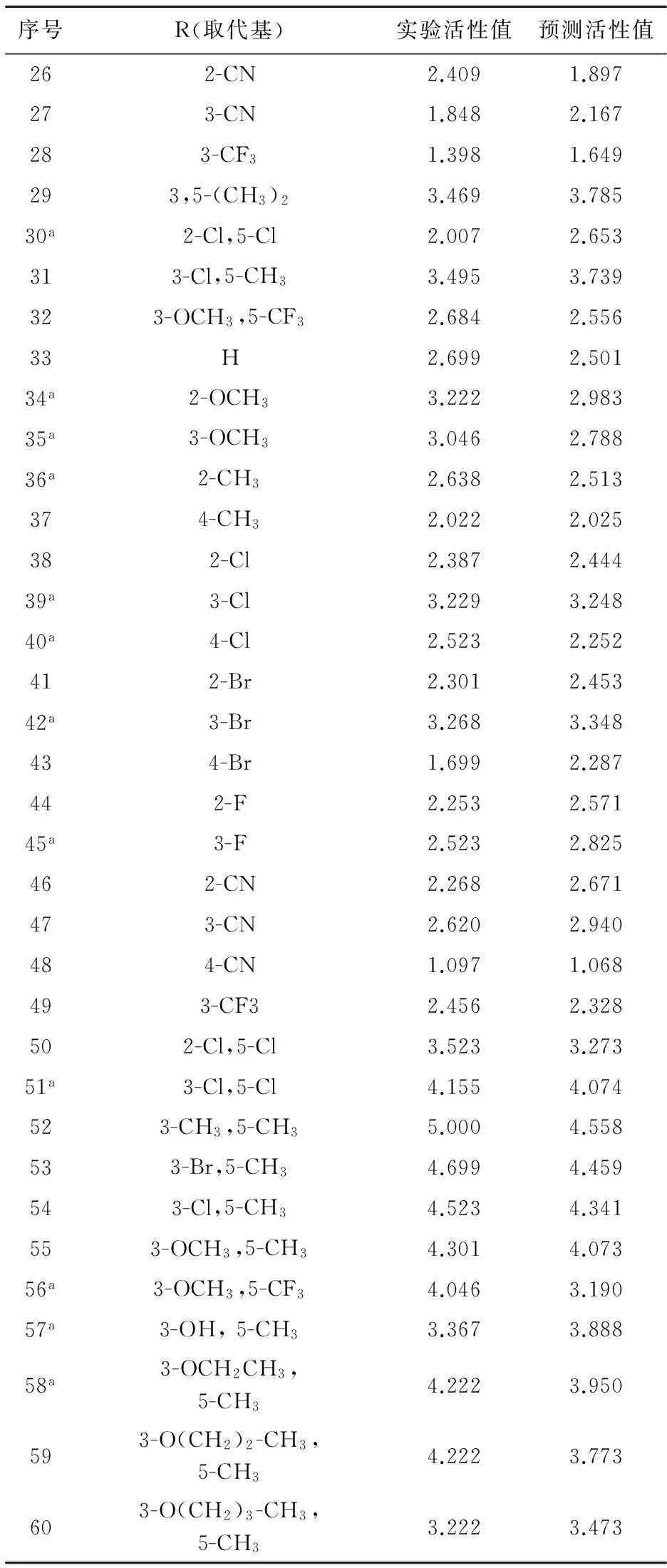
续表1
注:a为测试集
1.2 Topomer CoMFA模型建立
采用Tripos公司Sybyl 2.0-X软件包中的Sketch Molecule模块绘制出化合物的结构;对所有的化合物分子进行优化,优化过程采用Powell能量梯度法、Tripos力场等,能量收敛设定为0.05 kcal/mol,优化次数设定为1 000 次,分子荷载电荷为Gasteiger-Huckel电荷,其余参数以Sybyl 2.0-X默认值为准.
对42个训练集化合物的结构进行切割,以活性最高的52号分子为模板完成分子的切割,切割方式如图 1所示.选择以S为中心进行切割生成Ra和Rb基团,接着软件会对其余的分子结构自动识别并进行切割,最后对Ra和Rb基团周围的立体场和静电场进行计算.以立体场和静电场作为自变量,以化合物的活性作为因变量,采用偏最小二乘回归分析法建模生成3D-QSAR模型.采用留一法交互验证评价模型的内部预测能力.并利用建立的Topomer CoMFA模型对18个测试集化合物的活性进行预测,以此评价模型的外部预测能力.

图1 52号分子的切割方式
1.3 Topomer Search技术
采用Topomer Search技术可以从大量的化合物数据库中筛选出R基团(R-groups).其原理是:数据库中的分子被分割成片段,这些片段与训练集中的R-groups比较其Topomer相似性,通过阈值的 R-groups进一步打分,用模型预测其对活性的贡献.
本文利用建立的Topomer CoMFA模型对ZINC(2012)数据库中的Drug-like类(包含130 000个分子)进行筛选,以得到具有高活性贡献的Ra和Rb基团.其中,Topomer距离值设置为 185,其它参数以Sybyl 2.0-X默认值为准.
2结果与讨论
2.1 Topomer CoMFA建模结果与评价
由42个训练集化合物建立的Topomer CoMFA模型,其统计学参数见表2所示.由表2可知,拟合复相关系数r2、交互验证复相关系数q2分别为0.851、0.601.因此,本文所建立的Topomer CoMFA模型比较理想.
模型对18个测试集化合物预测的复相关系数qpred2为0.671.模型对42个训练集化合物和18个测试集化合物的预测活性值见表1所示.图2为训练集和测试集化合物活性实验值与预测值的线性回归图.从图2可以看出,该模型具有良好的拟合及预测能力.

表2 建模参数统计结果
注:aN:主成分数;r2: 拟合复相关系数;q2: 交互验证复相关系数;qpred2:外部验证复相关系数;SEE:标准估计偏差;SD: 拟合标准偏差;SDcv:交互验证标准偏差;F:F值.
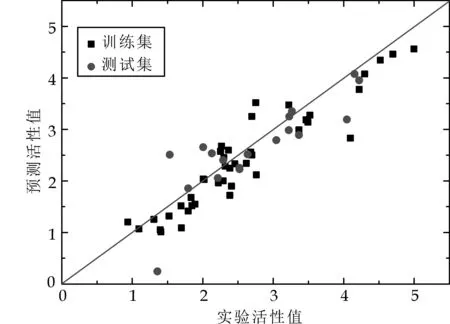
图2 60个化合物分子生物活性的实验值与预测值的线性回归图
2.2 Topomer CoMFA等势图分析
以52号分子为模板,Topomer CoMFA三维等势图见图3所示.图 3(a)和3(c)分别表示Ra和Rb基团的立体场等势图,绿色区域表示引入体积较大的取代基有利于活性的提高,黄色区域表示不宜引入体积较大的取代基;图3(b)与3(d)分别表示Ra和Rb基团的静电场等势图,红色区域表示引入负电性取代基有利于活性的提高,蓝色区域表示引入正电性取代基有利于活性的提高.
在图3(a)中,苯环的2位、4位附近有大片绿色区域,苯环的1位、3位附近有小片黄色区域;图3(b)中苯环的1、2、3位附近有蓝色区域,5、6位附近有大片红色区域;图3(c)中苯环的3位点和5位点处有大片绿色区域,4位点处有大块黄色区域;图3(d)中苯环的3位点、4位点和5位点处有大块蓝色区域.
综上所述,Ra基团中苯环的2位、4位引入体积大的基团有利于化合物活性的提高,1位和3位处适宜引入体积较小的取代基,并且在苯环的1、2、3位处引入带正电的基团有利于活性的提高,而在其5位、6位处引入带负电的基团有助于活性的提高.本文所引用的化合物体系都在苯环的1位、2位引入了带正电的氰基和氨基,而在其6位处则引入了带负电的硫基、亚磺酰基和磺酰基.
Rb基团中苯环的3位和5位点处适宜引入体积较大并带正电的基团,4位点处适宜引人体积较小并带正电的基团.例如51号分子与52号分子,51号分子在苯环的3位和5位处引入体积小并带负电的氯基,而52号分子引入体积比氯基大并带正电的甲基,因此52号分子的活性大于51号分子;又比如12号分子与14号分子,以带正电的氰基取代14号分子3位带负电的三氟甲基得到12号分子后活性明显增大.而6号分子的活性之所以比较低是因为分子的4位处引入了体积较大的甲基.

(a)Ra 立体等势图 (b)Ra 静电等势图
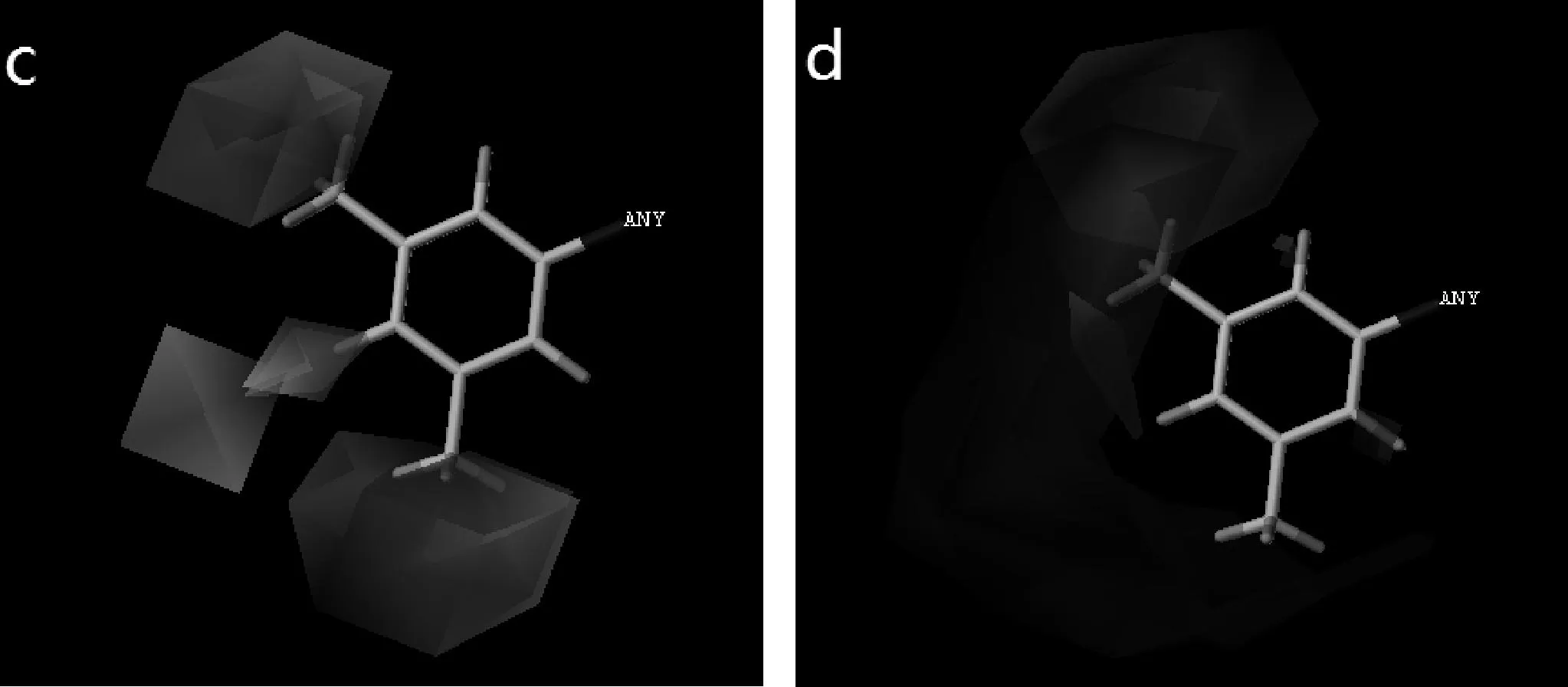
(c)Rb 立体等势图 (d)Rb 静电等势图图3 模型三维等势图
2.3 Topomer Search结果分析
Topomer Search的结果中主要包含两项:Topomer距离和R基团的活性贡献值.Topomer距离用来评价化合物结构的相似性程度;而R基团的活性贡献则是基于Topomer CoMFA模型对R基团活性值的预测打分.一般情况下,在设定的结构相似性程度范围内都会优先考虑贡献值的大小,以用于基于配体的分子优化设计以及虚拟筛选.
基于Topomer CoMFA模型,采用Topomer Search技术对ZINC(2012)数据库进行筛选,得到了5 000个Ra基团和5 000个Rb基团.以活性最高的52号分子为模板进行过滤,挑选出R基团活性贡献明显高于52号分子中的Ra和Rb活性贡献值的R基团,其中52号分子Ra贡献值为1.57,Rb贡献值为2.07.最终得到5个Ra基团和2个Rb基团.
2.4 分子设计
用最终筛选得到的Ra和Rb基团分别替换52号分子的Ra和Rb基团,得到了10个新化合物分子的结构.对新分子进行优化,优化方法同60个样本分子,利用建立的Topomer CoMFA模型对新化合物进行活性预测,10个新化合物的结构及其预测活性值见表3所示.由表3可知,有8个新设计的化合物预测活性值高于52号分子.
可以发现,新设计的分子Ra环基的6位都引入了带负电的磺酰基,因此活性有所增大.并且Rb环基的3位和5位引入了体积较大的基团,从而也提高了化合物的活性.例如,3号分子在此处引入体积较大的氨基和三氟甲基,所以它的活性较样本分子有所增大;而2号与7号分子的活性之所以比样本分子低,则是因为在Ra环的1位和3位处引入了体积较大的氨基和羰基,还有可能存在以下原因:一是,虽然单独考虑筛选出来的 Rb 基团贡献程度较大,但它与筛选出的Ra基团不匹配,所以导致活性偏低;二是,由于组合后的分子内部存在不利的相互作用而影响了活性的增大.以上解释都符合Topomer CoMFA模型等势图的分析.
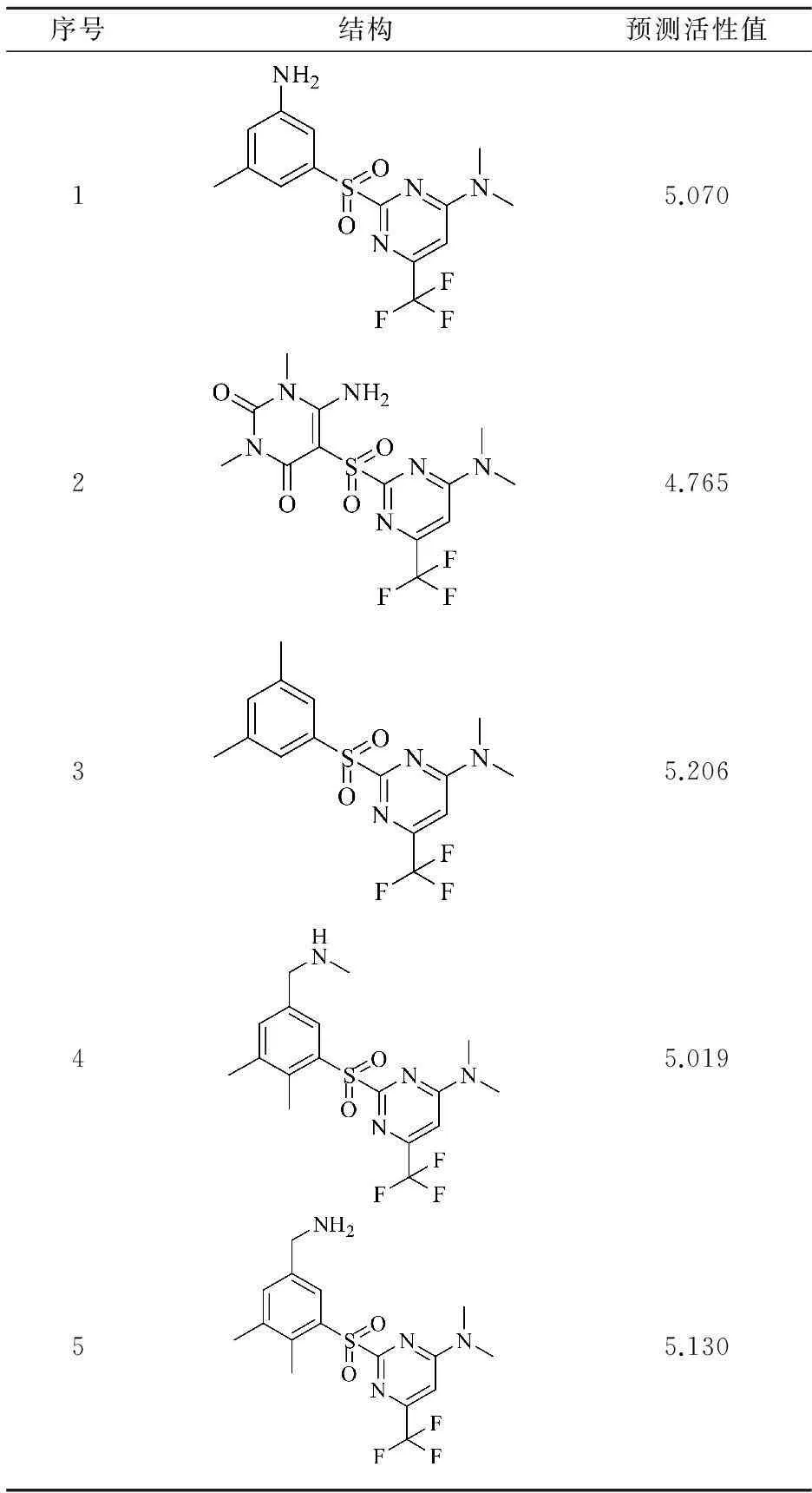
表3 新设计分子的结构及预测活性值

续表3
3结论
本文采用Topomer CoMFA方法对42个训练集2-氨基-6-磺酰苯甲腈衍生物分子进行了三维定量构效关系的研究,并利用所建立的模型对18个测试集化合物进行了活性预测.采用Topomer Search 技术对ZINC数据库进行了R基团的虚拟筛选,并设计出了10个新的化合物,其中8个具有比样本分子更高的活性.
结果表明,利用Topomer CoMFA方法能够建立有效的3D-QSAR模型,可以对未知化合物的生物活性进行预测,利用Topomer Search技术可以有效地从数据库中筛选出有高贡献的R基团,进一步设计出新的药物分子,从而为抗艾滋病新药设计提供新的侯选物.新设计的抑制剂对HIV-1病毒的抑制效果如何,尚需要进一步的实验验证.
参考文献
[1] Kunal Roy, J.Thomas Leonard.QSAR modeling of HIV-1 reverse transcriptase inhibitor 2-amino-6-arylsulfonylbenzonitriles and congeners using molecular connectivity and E-state parameters[J].Bioorganic & Medicinal Chemistry,2004,12(4):745-754.
[2] 姜大娥.探讨ELISA和化学发光法对血清中HIV-1/HIV-2 抗体、梅毒抗体和丙肝抗体的检测[J].中国卫生检验杂志,2012,22(3):543-548.
[3] 于国龙,刁丽梅,李杰,等.HIV-1与HIV-2交叉反应毒株的基因亚型分析[J].中国皮肤性病学杂志,2008,22(4):198-200.
[4] Ge Meng,Yang Liu,Aqun Zheng,et al.Design and synthesis of a new series of modified CH-diarylpyrimidines as drug-resistant HIV non-nucleoside reverse transcriptase inhibitors[J].European Journal of Medicinal Chemistry,2014,82:600-611.
[5] 徐永芳,林新勤,农全兴.艾滋病治疗研究现状[J].中国热带医学,2009,9(11):2 185-2 187.
[6] 龙亚秋.艾滋病治疗药物的研究进展[J].药学服务与研究,2007,7(6):401-407.
[7] Richard D.Cramer.Topomer CoMFA:A design methodology for rapid lead optimization[J].Journal of Medicinal Chemistry,2003,46(3):374-388.
[8] Richard D.Cramer,Phillip Cruz,Gunther Stahl,et al.Virtual screening for R-groups, including predicted pIC50 contributions, within large structural databases,using Topomer CoMFA[J].Journal of Chemical Information and Modeling,2008,48(11):2 180-2 195.
[9] H.Van De Waterbeemd.Chemometric methods in drug design[M].VCH:Weinheim,1995.
[10] Richard D.Cramer,David E.Patterson,Jeffrey D.Bunce J.Comparative molecular field analysis (CoMFA):Effect of shape on binding of steroids to carrier proteins[J].Journal of the American Chemical Society,1988,110(18):5 959-5 967.
[11] 许禄.化学计量学方法[M].北京:科学出版社,1995.
[12] 苗霞.以Tau蛋白为靶标治疗阿尔兹海默病的分子设计[D].重庆:重庆大学,2013.
[13] Richard D.Cramer,Farhad Soltanshahi,Robert Jilek,et al.All chem:Generating and searching 1020 synthetically accessible structures[J].Journal of Computer-Aided Molecular Design,2007,21(6):341-350.
[14] 刘永香,施海枫.基于Topomer CoMFA方法的姜黄素类化合物的三维定量构效关系研究[J].化工技术与开发,2014,43(2):36-38.
[15] Utpal Chandra De,Sudhan Debnath,Debanjan Sen.Studies on structural insight of 2-amino-6-arylsulfonylbenzonitrile derivatives as anti HIV agents[J].International Journal of Research in Pharmacy and Chemistry,2014,4(3):528-539.
[16] Rongjing Hu,Florent Barbault,Michel Delamar,et al.Receptor and ligand-based 3D-QSAR study for a series of non-nucleoside HIV-1 reverse transcriptase inhibitors[J].Bioorganic & Medicinal Chemistry,2009,17(6):2 400-2 409.
[17] Rongjing Hu,Jean Pierre Doucet,Michel Delamar,et al.QSAR models for 2-amino-6-arylsufonylbenzonitriles and congeners HIV-1 reverse transcriprase inhibitors based on linear and nonlinear regression methods[J].European Journal of Medicinal Chemistry,2009,44(5):2 158-2 171.
Application of an R-group search technology into molecular
design of 2-amino-6-arylsulfonylbenzonitrile derivatives
TONG Jian-bo1,2, BAI Min1, ZHAO Xiang1, CHANG Jia1
(1.College of Chemistry and Chemical Engineering, Shaanxi University of Science & Technology, Xi′an 710021, China; 2.Key Laboratory of Auxiliary Chemistry & Technology for Chemical Industry, Ministry of Education, Shaanxi University of Science & Technology, Xi′an 710021, China)
Abstract:Acquired Immunodeficiency Syndrome (AIDS) has been threatening human health,the study of anti-HIV drug design has become one of the important tasks of today's society.In this paper,a three-dimensional quantitative structure-activity relationship (3D-QSAR) study for 60 2-amino-6-arylsulfonylbenzonitrile derivatives was established using Topomer CoMFA.The multiple correlation coefficient of fitting,cross validation and external validation were 0.851,0.601 and 0.671,respectively.Topomer Search was used to search R groups from ZINC database.As the result,5 Ra groups and 2 Rb groups were selected.Finally,we designed 10 new compounds.The results indicated that Topomer CoMFA model had both favorable estimation stability and good predictive capability.Topomer Search technology could be effectively used to screen and design new compound,and had good predictive capability to guide the design of new Anti-HIV drugs.
Key words:Topomer CoMFA; 2-amino-6-arylsulfonylbenzonitrile derivatives; quantitative structure-activity relationship (QSAR); Topomer Search; new drug design
作者简介:仝建波(1975-),男,山西怀仁人,副教授,博士,研究方向:药物化学、食品化学、能源化工
基金项目:陕西省科技厅科技计划项目(2011K07-13); 陕西省教育厅专项科研计划项目(12JK0629,11JK0602);榆林市科技计划项目;陕西科技大学研究生创新基金项目
*收稿日期:2015-10-11
中图分类号:O641
文献标志码:A
*文章编号:1000-5811(2015)06-0067-05
