基于16S rDNA基因文库的黄河三角洲盐生植被土壤细菌群落多样性研究
2015-01-13姜爱霞郭笃发
张 玥,姜爱霞,郭笃发
(山东师范大学人口·资源与环境学院,山东济南 250014)
基于16S rDNA基因文库的黄河三角洲盐生植被土壤细菌群落多样性研究
张 玥,姜爱霞,郭笃发*
(山东师范大学人口·资源与环境学院,山东济南 250014)
[目的]研究黄河三角洲光板地及2种盐生植被(獐茅和罗布麻)下土壤细菌群落结构多样性,探究其与植被演替的关系。[方法]采用细菌16S rDNA基因文库方法,各文库挑选200个阳性克隆子进行测定。[结果]光板地、獐茅和罗布麻3个文库中分别得到121、150、155条有效序列。獐茅土壤细菌的Shannon指数和ACE指数最高。土壤中检测到变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)、酸杆菌门(Acidobacteria)等共8门。变形菌门在光板地、獐茅土壤和罗布麻土壤中的相对丰度分别为37.45%、51.09%、37.11%,拟杆菌门分别为33.02%、3.03%、4.47%,其他细菌相对丰度均较低。[结论]3种覆被类型下土壤细菌在种群组成与分布上差异明显,变形菌为优势菌群。当盐生植被处于不同演替阶段时,土壤细菌群落结构差别较大。
16S rDNA;黄河三角洲;细菌群落多样性;盐生植被
土壤微生物是生态系统的重要组成部分,其对土壤中几乎所有过程都起着直接或间接的作用。在土壤微生物中,细菌的数量和种类最多[1],其群落结构多样性的变化对土壤肥力、植被生长和污染物降解等具有重要影响,是评价自然及人为因素引起的土壤质量变化的重要指示因子[2]。
土壤细菌起初被认为呈全球随机性分布[3-4],后来的研究将多种影响土壤细菌群落结构多样性的因素归纳为自然和人为两大类[5],自然因素中的植物群落演替与土壤细菌群落结构的关系逐渐成为研究热点。研究表明,微生物群落结构变化与植被出现与否无关,表明植被对微生物的作用可能还与植被类型或植被演替阶段有关[6]。尽管植被群落与土壤微生物关系的研究取得了较大进展,但在滨海盐渍环境中,土壤细菌群落多样性的变化与盐生植被演替的关系尚不明晰。
笔者采用近年来在细菌多样性领域应用广泛的16S rDNA克隆文库技术,研究黄河三角洲盐生植被土壤细菌群落多样性,探究其与植被演替的关系,为进一步研究黄河三角洲自然保护区湿地生态系统提供理论依据。
1 材料与方法
1.1 土样采集在黄河三角洲研究区内沿东西向路线(横跨现代三角洲和近代三角洲),依照盐生植被正向演替顺序,于2012年6月选择光板地、一种强耐盐植被獐茅、一种轻耐盐植被罗布麻,每个植被群落取3个样地,每个样地采用对角线五点采样法采集土壤样品。
采集0~20 cm深的土壤样品,并混合均匀,除去较大的根系等杂物,分成2份,每份约200 g。一份土样封于灭菌袋中,保存在4 ℃条件下,用于微生物培养和土壤理化性质分析;另一份土样放入液氮罐带回实验室,保存在-80 ℃条件下,用于分子生物学研究。
1.2 DNA提取使用Omega DNA提取试剂盒对土壤样品DNA进行提取,具体操作步骤见试剂盒操作说明书。
1.3 细菌16S rDNA 的PCR扩增PCR扩增所用引物为:27F(5′-AGAGTTTGATCCTGGCTCAG-3′)、1525R (5′-AGA-AAGGAGGTGATCCAGCC-3′)。PCR反应体系的各组分组成为:引物(10 pmol/L)各0.4 μl ,DNA模版1.0 ng,MgCl2(2.5 mmol/L) 0.3 μl,10×Buffer 2.0 μl,dNTP(2.5 mmol/L)0.4 μl,DNA聚合酶0.2 μl,Milli-Q Water补至20 μl。PCR扩增反应条件:94 ℃预变性5 min;94 ℃变性30 s,55 ℃退火1 min,72 ℃延伸2 min,30个循环;72 ℃延伸10 min。PCR产物采用荧光染料作为标记,用0.8%(W/V)的琼脂糖凝胶在1×TAE的缓冲液中进行电泳测定。
1.4 扩增产物的克隆和测序使用试剂盒(Tiangen Biotech Beijing)回收16S rDNA片断并进行纯化。将纯化产物连接至pMD 19-T Vector载体上,再转入大肠杆菌(Escherichiacoli)感受态细胞中。在含有氨苄青霉素LB培养基平板上分别涂布,37 ℃下培养过夜。利用T7和SP6进行菌落PCR。PCR扩增后检验,挑选插入目的片断的阳性克隆子,送山东农业科学院生物信息技术服务中心测序。
1.5 序列分析及发育树的构建将测序所得序列在BLASTN中进行同源性比对,挑选与序列亲缘关系最相似序列,使用CHECK-CHIMERA软件分析检查,去除嵌合及怪异序列。利用ClustalW2软件将测定的序列和部分网上下载的16S rDNA相似序列进行多重分析比对,找出可操作分类单元(OTU)。使用Mega5.0软件中的邻接法建构系统发育树。
1.6 多样性指数计算使用Mothur软件进行土壤细菌α多样性分析,计算α多样性指数,包括丰富度指数(Richness)、覆盖率(Coverage)、香农指数(Shannon)、ACE指数、Chao1指数。
2 结果与分析
2.1 土壤理化性质黄河三角洲土壤可溶性盐分主要为NaCl,土壤电导率与盐分含量能用线性进行模拟[7],因此可用土壤电导率表征土壤含盐量。由表1可知,3种盐生植被下土壤1∶5土水比浸提液的电导率在0.04~1.95 dS/m。光板地、獐茅地为海积潮湿正常盐成土,其中光板地盐化程度比獐茅地高很多,罗布麻地为弱盐淡色潮湿雏形土;根据国际土壤质地分类制,光板地属于粉砂壤土,獐茅地属于粉砂黏壤土。有机质、全氮、碱解氮在獐茅和罗布麻土壤中的含量均高于光板地,罗布麻地速效磷含量高于光板地和獐茅地,差异明显。由此可知,土壤速效磷随盐生植被正向演替有不断升高的趋势。光板地的温度高于其他覆被类型。
表1 不同覆被类型土壤理化性质
2.2 土壤细菌测序深度及多样性分析由图1可知,OTU数目随序列数增多而增加,但增幅逐渐变小,曲线趋于平缓,但仍未达到饱和,表明挑取的克隆数能基本反映土壤样品中细菌群落的结构组成。由表2可知,3种覆被类型下土壤细菌覆盖率在52.67%~61.67%,表明黄河三角洲土壤细菌具有较高的多样性。獐茅土壤Shannon指数最高,为8.52,说明獐茅土壤细菌具有较高的多样性,光板地Shannon指数最低,为7.19。ACE指数和Chao1指数在3种土壤中差异明显,獐茅土壤ACE指数最高,为31 554.68,獐茅土壤ACE指数和Chao1指数均最高,分别为3 155.68和18 806.41。相关性统计分析表明,土壤理化性质与各多样性指数无显著相关关系。
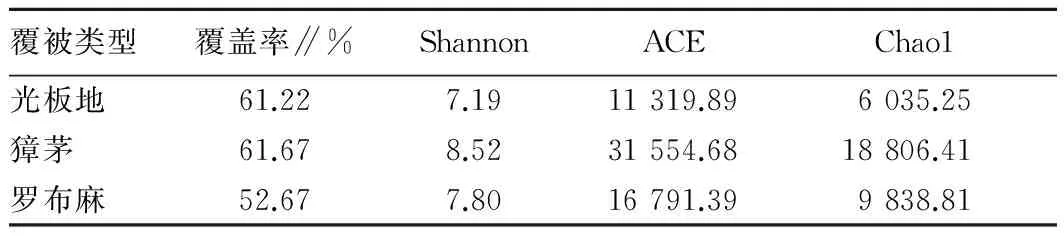
表2 不同土壤细菌多样性指数
2.3 土壤细菌16S rDNA序列分析通过Mothur分析,将相似性大于97%的序列归为同一种OTU,得到有效序列和OTU分析结果:光板地有效序列121个,OTU 72个;獐茅有效序列150个,OTU 83个;罗布麻有效序列155个,OTU 88个。
对系统进化树进行分析,发现细菌序列主要归属于8个门类:变形菌门(Proteobacteria),拟杆菌门(Bacteroidetes),异常球菌-栖热菌门(Deinococcus-Thermus),酸杆菌门(Acidobacteria),绿弯菌门(Chloroflexi),厚壁菌门(Firmicutes),疣微菌门(Verrucomicrobia)和放线菌门(Actinobacteria)。8种土壤细菌门在3种土壤中的分布情况见图2。由图2可知,变形菌门在3种土壤中相对丰度为37.11%~51.09%,为优势菌群,经相关性分析,δ-变形菌(δ-Proteobacteria)与土壤全氮相关系数为0.89 (P<0.05),显著相关;拟杆菌门相对丰度次于变形菌门,且在3种土壤中的分布差异明显,为3.03%~33.02%,光板地中相对丰度最高,经相关性分析,拟杆菌门与土壤电导率(含盐量)相关系数为0.99 (P<0.01),极显著相关;酸杆菌门在3种土壤中相对丰度为0.21%~23.87%,在罗布麻土壤中相对丰度最高;放线菌门在3种土壤中相对丰度为2.32%~5.90%,在獐茅土壤中相对丰度最高;厚壁菌门在3种土壤中相对丰度为1.43%~4.22%,在罗布麻土壤中相对丰度最高,经相关性分析,厚壁菌门与土壤含水率相关系数为0.94 (P<0.05),显著相关;绿弯菌门在3种土壤中相对丰度为0.81%~3.43%,在獐茅土壤中相对丰度最高;疣微菌门在3种土壤中相对丰度为0.37%~1.59%,在罗布麻土壤中相对丰度最高;异常球菌-栖热菌门只在光板地中检测到,相对丰度为0.23%。
2.3.1变形菌门。在獐茅土壤中相对丰度最高,为51.09%,有40个OTU。α-变形菌(α-Proteobacteria)含量最高,在獐茅土壤中相对丰度为14.67%,有17个OTU,其中12个属于Rhodospirillales,3个属于Rhizobiales,2个属于Rhodospirillales;γ-变形菌(γ-Proteobacteria)相对丰度次之,为13.33%,有7个OTU,3个属于绿脓杆菌(Pseudomonas),1个属于硫发菌目(Thiotrichales),1个属于海螺菌目(Oceanospirillales),1个属于交替单胞菌目(Alteromonadales);β-变形菌(β-Proteobacteria)相对丰度为2.67%,有4个OTU,均属于伯克氏菌目(Burkholderiales);δ-变形菌(δ-Proteobacteria) 相对丰度最低,为2.00%,有3个OTU,1个OTU属于互营杆菌目(Syntrophobacterales),1个属于脱硫菌目(Desulfobacterales),1个OTU属于粘球菌目(Myxococcales)。
在光板地中相对丰度次之,为37.45%,有16个OTU。δ-变形菌含量最高,在光板地中相对丰度为5.79%,有7个OTU,其中6个OTU属于脱硫菌目,1个属于粘球菌目;γ-变形菌相对丰度次之,为4.96%,有5个OTU;α-变形菌相对丰度为1.65%;β-变形菌相对丰度为1.65%,有1个OTU,均属于伯克氏菌目丛毛单胞菌科丛毛单胞菌属(Comamonas)。
在罗布麻土壤中相对丰度最低,为37.11%,有13个OTU。γ-变形菌在罗布麻土壤中相对丰度最高,为9.68%,有4个OTU;δ-变形菌相对丰度次之,为3.23%,有5个OTU,全部属于粘球菌目,β-变形菌相对丰度最低,为1.94%,有3个OTU,均属于伯克氏菌目。
2.3.2拟杆菌门。拟杆菌门在光板地中相对丰度最高,为32.02%。有17个克隆体为拟杆菌纲(Bacteroidetes),10个克隆体为海洋滑行菌属(Marinilabilia),4个克隆体属于鞘脂杆菌纲(Sphingobacteria);在罗布麻土壤中相对丰度次之,为4.47%,有13个OTU,全部为鞘脂杆菌纲;在獐茅土壤中相对丰度最低,为3.03%,有3个OTU,全部为鞘脂杆菌纲。
2.3.3酸杆菌门。在罗布麻土壤中相对丰度最高,为16.65%,有30个OTU,其中包含Gp4 6个OTU,Gp9 3个OTU,Gp5 3个OTU,Gp7 2个OTU,Gp26、Gp17、Gp1、Gp3各1个OTU;在獐茅地土壤中相对丰度次之,为7.01%,有5个OTU,分别属于Gp26、Gp21、Gp4、Gp10;在光板地中相对丰度最低,只有0.21%,有2个OTU,分别属于Gp3、Gp10。经相关性分析,Gp7、Gp15与土壤有效磷相关系数均为0.90 (P<0.05),显著相关。
2.4 土壤细菌群落结构比较由图3可知,光板地与属于重盐土壤的獐茅土壤细菌及属于轻盐土壤的罗布麻土壤细菌间共有的OTU序列为16个,分别占三者OTU总数的66.67%、53.33%和80.00%;光板地与属于重盐土壤的獐茅土壤细菌间共有的OTU序列为5个,分别占两者OTU总数的20.83%和16.67%;光板地与属于轻盐土壤的罗布麻土壤细菌间共有的OTU序列为0个;属于重盐土壤的獐茅土壤与属于轻盐土壤的罗布麻土壤细菌间共有的OTU序列为3个,分别占两者OTU总数的10.00%和15.00%。由此可知,当盐生植被在不同演替阶段时,土壤细菌群落结构差别较大。
3 结论与讨论
该研究利用细菌16S rDNA基因文库克隆测序技术,对黄河三角洲不同演替阶段的代表性盐生植被:光板地、獐茅和罗布麻的土壤细菌群落结构多样性进行分析,进而明确其与植被演替的关系。变形菌门在3种植被土壤中为优势菌群,在属于重盐土壤的獐茅土壤中相对丰度最大,这与以往利用细菌16S rDNA克隆文库方法对土壤样品进行研究所得结论基本一致。Tait等[8]对美国马萨诸塞州礁湖的研究、Schloss等[9]对农田土壤样品细菌的研究、Axelrood等[10]对英国哥伦比亚森林土壤的研究,均发现变形菌门为土壤中的优势菌群。拟杆菌门在3种土壤中相对丰度次于变形菌门,在光板地中相对丰度最高,其余2种土壤中相对丰度均低,且与土壤电导率(含盐量)呈极显著相关关系,表明其分布可能与土壤盐分有关。
酸杆菌门是最近被划分出的一门细菌,多为嗜酸菌,目前对其研究较少,但已有研究表明酸杆菌门广泛存在于土壤中,且为优势菌种[10]。酸杆菌门主要分布于陆地、海洋沉积物和活性淤泥中,酸性土壤有利于其生长,该研究中,酸杆菌门在光板地、重盐土壤(獐茅土壤)中的相对丰度明显小于其在轻盐土壤(罗布麻土壤)中。关于酸杆菌门中的Gp7、Gp15与土壤有效磷显著相关的原因,有待进一步研究。
异常球菌-栖热菌门包括一些能抵抗严酷环境的球状细菌,有较强的耐热、抗辐射、抗砷能力[11]。该研究只在光板地中检测到异常球菌-栖热菌门的出现,占有较大比重,且与土壤电导率(含盐量)呈极显著相关关系,这或许说明其也有高强度的耐盐性。Saccharospirillum在轻盐土壤(罗布麻土壤)中丰度大于其他类型土壤,今后可对其进行深入研究,探讨将其作为黄河三角洲脱盐演化指示性细菌的可能性。
许多细菌类群的分布并未发现有规律的演替变化,如弯菌门和疣微菌门,呈现一定的随机性,它们的分布除受土壤含盐量影响外,可能还与土壤植被类型以及人为影响等多种因素的共同作用有关[12]。
目前,对环境样品16S rDNA克隆文库的分析,通常随机选取50~100个克隆子探讨其细菌类群和多样性[13]。由于克隆文库较小,不能包含样品所有细菌的种类,难以获得整个细菌群落的所有信息。该研究每个克隆文库都选取了200个克隆子,基本能够科学反映所研究样品的优势细菌类群和多样性。
[1] 林先贵,胡君利.土壤微生物多样性的科学内涵及其生态服务功能[J].土壤学报,2008,45(5):892-900.
[2] 蒋婧,宋明华.植物与土壤微生物在调控生态系统养分循环中的作用[J].植物生态学报,2010,34(8):979-988.
[3] O’MALLEY M A.The nineteenth century roots of everything is everywhere[J].Nature Reviews Microbiology,2007,5(8):647-651.
[4] 刘银银,李峰,孙庆业,等.湿地生态系统土壤微生物研究进展[J].应用与环境生物学报,2013,19(3):547-552.
[5] PERALTA A L,MATTHEWS J W,KENT A D.Microbial community structure and denitrification in a wetland mitigation bank[J].Appl Environ Microbiol,2010,76(13):4207-4215.
[6] ANDERSEN R,GRASSET L,THORMANN M N,et al.Changes in microbial community structure and function following Sphagnum peatland restoration[J].Soil Biology and Biochemistry,2010,42:291-301.
[7] 翁永玲,官鹏.黄河三角洲盐渍土盐分特征研究[J].南京大学学报:自然科学版,2006,42(6):602-610.
[8] TAIT E,CARMAN M,SIEVERT S M.Phylogenetic diversity of bacteria associated with ascidians in Eel Pond (Woods Hole,Massachusetts,USA)[J].Journal of Experimental Marine Biology and Ecology,2007,342(1):138-146.
[9] SCHLOSS P D,HANDELSMAN J.Status of the microbialcensus[J].Microbiology and Molecular Biology Reviews,2004,68(4):686-691.
[10] AXELROOD P E,CHOW M L,RADOMSKI C C,et al.Molecular characterization of bacterial diversity from British Columbia forest soils subjected to disturbance[J].Canadian Journal of Microbiology,2002,48(7):655-674.
[11] 张玉琴,李文均,郝涤非,等.异常球菌属的分类及应用研究进展[J].微生物学通报,2006,33(6):133-137.
[12] 张瑞娟,李华,林勤保,等.土壤微生物群落表征中磷脂脂肪酸(PLFA)方法研究进展[J].山西农业科学,2011,39(9):1020-1024.
[13] 陈彦闯,辛明秀.用于分析微生物种类组成的微生物生态学研究方法[J].微生物学杂志,2009,29(4):79-83.
Analysis of Saline Vegetation Diversity of Soil Bacterial Community in the Yellow River Delta Based on 16S rDNA Gene Library
ZHANG Yue, JANG Ai-xia, GUO Du-fa*
(College of Population, Resources and Environment, Shandong Normal University, Jinan, Shandong 250014)
[Objective]The aim of the study was to investigate the soil bacterial community structure of three kinds of saline vegetation (Bare Board, Angiospermae andA.venetum) in the Yellow River Delta, and reveal its relationship with saline vegetation succession. [Method]Phylogenetic trees were constructed by the use of bacterial 16S rDNA gene library, for each of the five libraries constructed, 200 positive clones were picked randomly and sequenced, and the data were statistically analyzed. [Result]A total of 121, 150 and 155 valid sequences were obtained from bare board, Angiospermae and A. venetum. The Angiospermae soil had the highest Shannon index and ACE index. Bacteria belonging to 8 phyla were identified, like Proteobacteria, Bacteroidetes, Acidobacteria and so on. Among them, Proteobacteria accounted for 37.45%, 51.09%, 37.11% form bare board, Angiospermae andA. venetum soil, Bacteroidetes accounted for 33.02%, 3.03%, 4.47%, while the other bacterial phyla each accounted less. [Conclusion]The soil bacterial community structure of the three soil bacteria differed significantly, and Proteobacteria was the advantage microflora. The community structure of soil bacteria had significantly differences when the saline vegetation succession at the different stage.
16S rDNA; The Yellow River Delta; Bacterial community diversity; Saline vegetation
山东省自然科学基金项目(2012ZRB019DW)。
张玥(1990-),女,山东烟台人,硕士研究生,研究方向:环境微生物。*通讯作者。
2015-03-25
S 182
A
0517-6611(2015)13-055-03
