纤锌矿结构ZnO、AlN、GaN自发极化及压电系数的第一性原理计算
2014-12-31牛海波陈光德耶红刚
牛海波,陈光德,耶红刚
(西安交通大学 理学院,陕西 西安 710049)
宽禁带半导体材料具有禁带宽度大、电子漂移饱和速度高、介电常数小、导电性能好的特点,这些优越的性质使其在深紫外发光及光电探测器件、高温高频大功率电子器件、微波功率器件等领域应用中有着潜在的巨大前景,非常适用于制作抗辐射、高频、大功率和高密度集成的电子器件[1],已经成为目前半导体研究领域中的热点.北京大学、西安电子科技大学等高校已经建立了宽禁带半导体研究中心.宽禁带半导体通常具有六方纤锌矿结构,由于其c轴方向存在非中心对称的结构,导致产生较大的自发极化和压电极化,发生很多中心对称结构半导体所缺少的性质如压电效应、热电效应等.极化场对宽禁带半导体的性质具有重要影响,王中林课题组正是根据ZnO纳米线阵列的压电特性,发明了纳米发电机,开创了一个新的研究领域[2].另一方面,极化效应引起的内建电场可达到MV/cm,对载流子分布、电场分布具有显著的作用,导致半导体中能带弯曲,降低发光效率,对器件的光电性能产生重要的影响[3-5].因此分析认识纤锌矿半导体中的极化场性质,进而对其加以调控,扬长避短,对于改进和提高光电设备性能,制造新型光电仪器具有重要的现实意义.
本文以目前3种具有广阔应用前景的纤锌矿结构宽禁带半导体ZnO、GaN、AlN(室温下禁带宽度分别为3.37eV、3.36eV、6.2eV)为研究对象,以现代极化理论为基础并结合第一性原理计算,通过构建一种结构简单、直观的计算模型,分别采用Berry phase和最大局域化Wannier函数方法,对这3种半导体中的自发极化及压电系数进行计算,分析自发极化产生的原因,并对比了这两种方法在分析极化问题中的优缺点.
1 计算理论
1.1 自发极化
分析计算物质结构中的电子极化传统上采用的是 Clausius-Mossotti(CM)模型[6],这种方法对于电子局域分布的物质如NaCl等较为适用,但是对于电子非局域分布的结构如宽禁带半导体,则会带来很大的困扰,表现在计算周期的选取会导致结果的不一致.为了解决上述问题,Vanderbilt于1993年提出了现代极化理论[7],其核心思想是不直接研究极化值,而以具有物理意义的极化的改变量为研究对象,当系统绝热缓慢地由状态λ1变为状态λ2时,极化量的变化可由产生的Berry phase相计算得到.极化量的改变可以表示为

半导体中的极化P可以表示成离子极化Pion与电子极化Pe之和,即

对于离子极化的计算,由于离子是局域化分布在结构中,因此可采用下式计算:

式中qi为离子所带电量,ri为离子位置.
Pe的计算方法有两种,分别是Berry phase方法[7]和最大局域化 Wannier函数方法[8].对于 Berry phase方法,Pe采用下式计算:
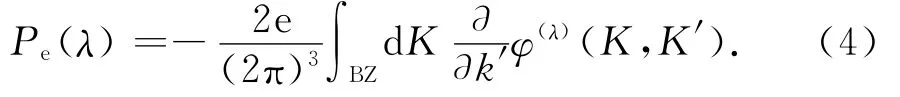
式中:积分区间为第一布里渊区,φ(λ)为占据态布洛赫函数的几何量子相位,即Berry phase相,计算系统绝热缓慢变化过程中产生的相位是该方法的关键所在.
最大局域化Wannier函数方法采用正交且空间局域的Wannier基函数,通过对电子分布进行处理,得到各电子占据态的Wannier中心,将非局域分布的价电荷都认为集中在局域分布的Wannier中心上,因此可直观地将Wannier中心看作点电荷,每个Wannier中心带电量为-2e,整个晶体从而可以分成局域分布的带正电的离子和带负电的Wannier中心,以此为基础进而分析电子结构.在这种方法中,Pe可简单表示为
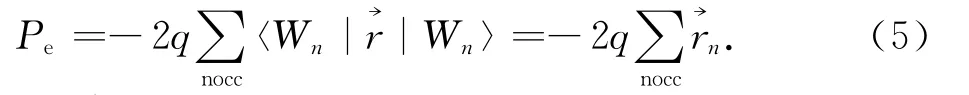
式中→rn为Wannier中心位置与Berry phase方法相比,Wannier函数方法计算得到的结果与CM模型相类似,给出的物理意义比较清楚直观,更易于理解.
1.2 压电系数
纤锌矿半导体外延层一般沿[0001]方向生长,而且主要是由于该方向上存在非对称结构,导致产生较大的自发极化,因此本文主要研究[0001]方向即沿c轴的自发极化与压电极化,与该方向相关的压电系数有3个,即e33、e31及e32,分别反映了c轴、a轴及b轴施加应力下极化的变化,由于纤锌矿结构的对称关系,其中e31=e32.压电系数采用下式进行计算[9-10]:
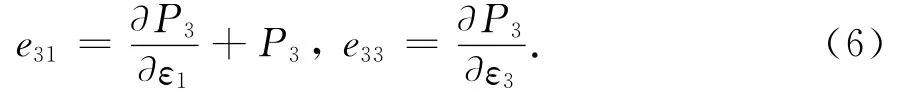
式中P3是c轴方向的极化值,ε1=(a-a0)/a0和ε3= (c-c0)/c0分别表示a轴和c轴上施加的应变,a0和c0分别是未加应变时晶体的晶格参数,在本文中施加的应变限制在±1%范围.
2 计算模型及方法
2.1 计算模型
由于现代极化理论计算的是极化的变化量,因此要计算纤锌矿结构ZnO、GaN、AlN的自发极化,必须要建立自发极化为零的参照结构,纤锌矿结构与参照结构的极化值之差才是所求的自发极化,闪锌矿结构由于中心对称一般被用作参照模型.不同于其他研究中建立较大的参照模型,我们建立了一种简单直观的模型,计算起来较为简便,以ZnO为例,本文是在纤锌矿ZnO原胞基础上,在c基矢方向扩展3个单位得到1×1×3的纤锌矿结构的超晶胞,按照ABAB顺序堆垛而成,如图1b所示.然后根据闪锌矿结构特点,分别移动Zn原子和O原子,按照ABCABC顺序堆垛构建了闪锌矿结构的参照模型,如图1a所示.每种模型分别包含6个Zn原子和O原子,且体积相同,晶格参数也保持不变.当由闪锌矿参照结构缓慢绝热地变化为纤锌矿结构,即可根据现代极化理论进行自发极化的计算.
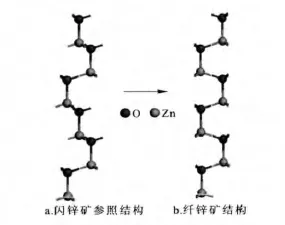
图1 ZnO闪锌矿参考结构(a)及纤锌矿结构(b)Fig.1 Zinc blend reference(a)and wurtzite structures of ZnO(b)
2.2 计算方法
我们采用开源软件Quantum-ESPRESSO[11]进行计算,它是利用第一性原理,以密度泛函理论和分子动力学理论为基础的应用广泛的软件,包含以现代极化理论为基础的Berry phase方法.最大局域化Wannier函数方法利用 Wannier90[12]程序包计算,Wannier90作为一种后处理程序,实现了和Quantum-ESPRESSO的无缝连接.计算中采用广义梯度近似(GGA)的PBE来处理电子之间的交互关联能,选择的赝势为Vanderbilt超软赝势,由于自发极化与晶体结构参数紧密相关,因此在参数设置中,平面波截断能设为40Ry,选取9×9×2的Monkorst-ParkK点对全Brillouin区求和.对于Berry phase方法,在计算c轴方向极化时K点加密,采用9×9×7.总能变化收敛的标准为1.0×10-6eV,原子间的相互作用力收敛标准为0.05eV/nm.
3 计算结果及讨论
由于自发极化对晶格参数非常敏感,在计算自发极化前我们首先对3种半导体的原胞进行仔细的优化,然后对以此为基础构建的闪锌矿结构的参照模型进行优化,结果见表1,其中参数u为纤锌矿结构中平行于c轴方向的键长与晶格参数c的比值,反映了c方向原子层的间距.
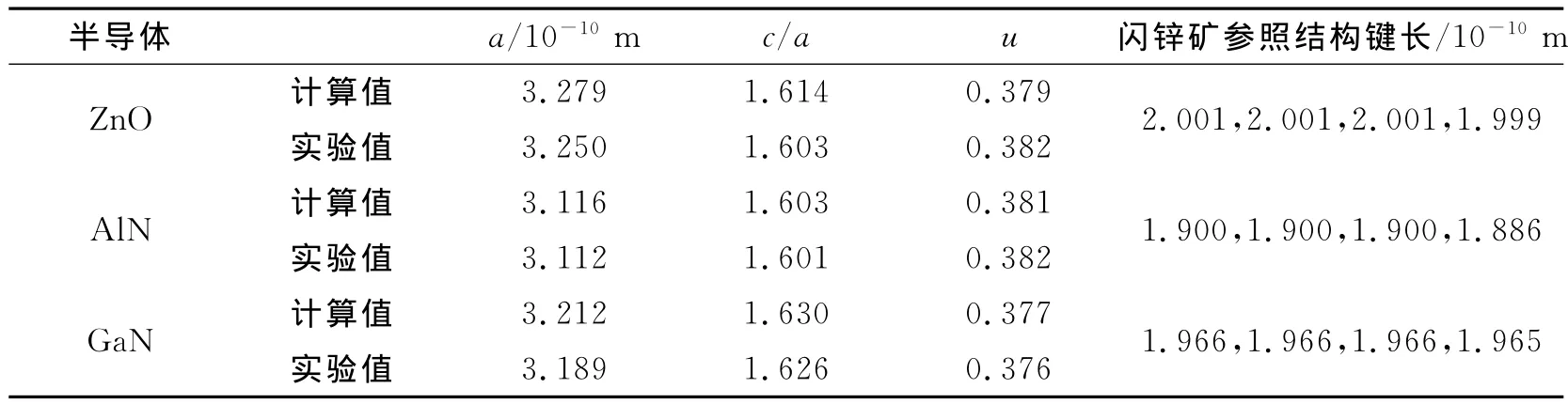
表1 结构优化后3种纤锌矿结构晶体原胞晶格参数及对应的闪锌矿参照结构键长Tab.1 Lattice parameters of the relaxed wurtzite ZnO,AlN and GaN primitive cell and the bond length of the zinc blend reference structure
从表1中可以看到计算得到的晶格参数与实验值非常接近,最大的相对变化也只有0.89%.计算值稍大于实验值,这与交换关联能使用GGA近似有关,GGA近似比较适合电子密度不均匀的体系,一般情况下采用GGA计算得到的晶格常数均会轻微增加.晶格参数c/a和u均偏离纤锌矿结构理想值1.633和0.375,这是导致晶体具有自发极化的原因.优化后的闪锌矿参照结构的4个键长非常接近,最大的相对变化为0.73%,说明构建的参照结构符合闪锌矿的正四面体结构特征.
3.1 自发极化计算
3.1.1 Berry phase方法 利用Berry phase方法,分别对闪锌矿参照结构和纤锌矿结构进行了计算,得到两种结构下的极化值,然后通过二者之差计算出自发极化,由表2可以看出,AlN、GaN的自发极化计算值与其他文献结果非常接近,特别是最近Jonas[13]利用实验得出 GaN的自发极化为-0.029 C/m2,证明了我们的计算模型及计算方法的正确性.ZnO的计算值接近于文献[14]而与文献[15]差别比较大,这主要是因为与其他文献使用的交换关联能及晶格参数存在差异导致,特别是u值不同,因为自发极化对u值很敏感,我们利用文献[15]给出的u值进行了计算,最后得出的自发极化为-0.036 C/m2,靠近文献[15]的结果.同时可以看出AlN的自发极化在三者中最大,这是因为AlN的结构参数c/a和u偏离理想值最大,非中心对称的程度也最大.这与文献[15]得出的结果一致.
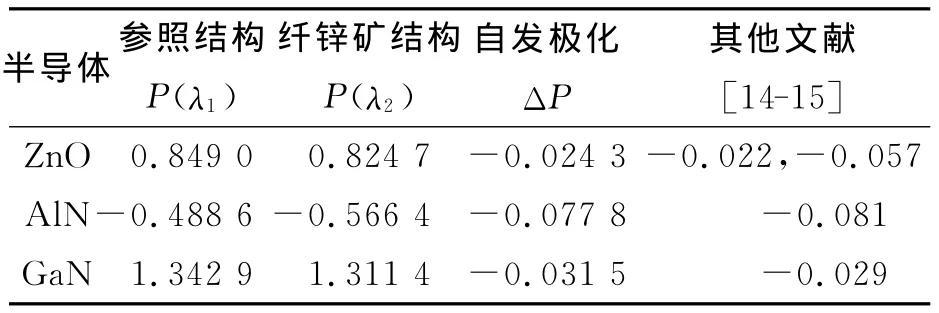
表2 参照结构、纤锌矿结构极化计算值及自发极化Tab.2 The calculated polarization of reference and wurtzite structures C/m2
3.1.2 Wannier函数方法 利用 Quantum-ESPRESSO软件进行3种半导体的自洽场和非自洽场计算后,再使用Wannier90软件包对前面步骤中得出的电子密度进行处理可得到Wannier中心.由于闪锌矿及纤锌矿结构中各原子间以sp3杂化成键,根据电子占据态,每一个成键轨道对应一个Wannier中心,因此参考结构和纤锌矿结构模型中每个都含有24个Wannier中心,根据式(3)可以得出各半导体的离子极化、电子极化,再根据式(1)二者做差计算得出自发极化,结果见表3.
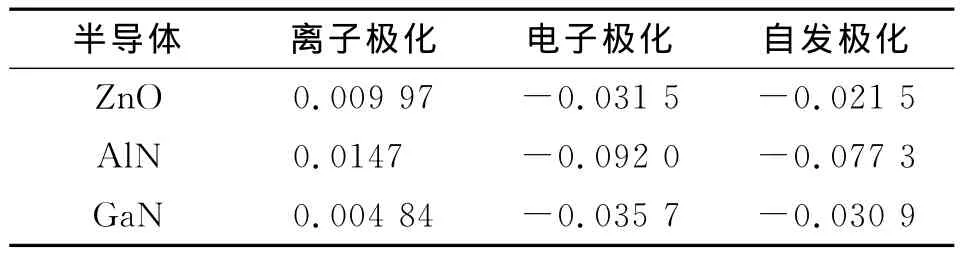
表3 利用Wannier90计算得到的3种半导体的各项极化Tab.3 The ionic,electric and spontaneous polarization of the three semiconductors calculated with Wannier90 C/m2
计算得到的自发极化与用Berry phase方法得到的非常接近,且电子极化值远大于离子极化,说明3种半导体的自发极化主要来自于电子极化的贡献,由于 N原子的电负性(3.04)比 Al原子(1.61)、Ga原子(1.81)大很多,成键时N原子能够强烈吸引与其键合的原子的电子云,使得三族氮化物的共价键同时具有很强的离子性.ZnO晶体与三族氮化物具有相似的性质,这从其Wannier中心分布可以明显看出.
图2所示为计算得到的3种纤锌矿结构半导体的Wannier中心,可以清楚的看到Wannier中心非常靠近O原子和N原子,说明结构中的共价键具有较强的离子性,根据Hazem[16]的理论,我们计算了3种半导体中键的离子性,分别为ZnO:0.802,AlN:0.764,GaN:0.731,远大于0.500的临界指标.以上分析说明3种半导体强烈的自发极化与键的离子性有着紧密的联系,由于表3中计算的是沿着晶体c轴的极化值,我们也利用Wannier函数方法计算了a、b轴两个方向的极化值,结果趋于0,说明纤锌矿结构中自发极化主要沿着c轴产生;另一方面,在图2中,c轴方向的Wannier中心位置要比其他3个更接近于阳离子,导致沿c轴正负离子中心不重合,产生自发极化.
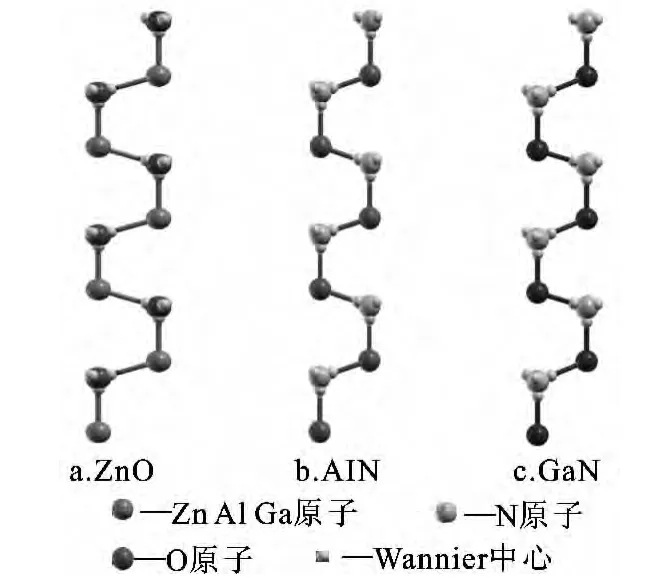
图2 纤锌矿结构ZnO、AlN、GaN中Wannier中心分布Fig.2 Wannier centers of wurtzite ZnO,AlN and GaN
3.2 压电系数计算
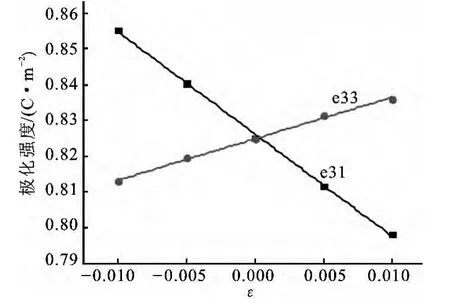
图3 不同应变条件下ZnO极化值Fig.3 Polarization of ZnO as a function of strain
沿c轴施加不同程度的应变,计算该条件下c方向上的极化值.以ZnO为例,如图3所示,在微小形变内施加的应变与极化值之间存在良好的线性关系,根据式(6),计算得到了3种半导体的压电系数,结果见表4.计算结果与参考值[15]符合的很好,从表3和表4可以看出,AlN的自发极化及压电系数是3种半导体中最高的,其值是经典的铁电体钙钛矿自发极化的1/3,AlN的自发极化甚至超过了其余两种半导体的两倍.较强的自发极化和压电极化,显示AlN在压电领域有着更广泛的应用前景.
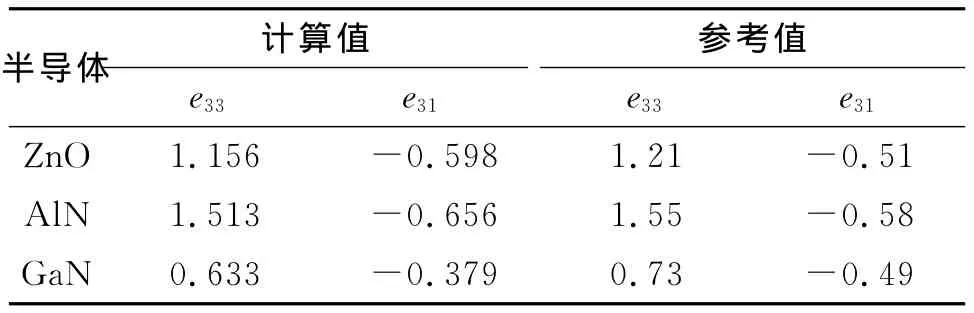
表4 ZnO、AlN及GaN压电系数Tab.4 Piezoelectric constants of ZnO,AlN and GaN C/m2
4 结论
根据现代极化理论并结合第一性原理计算,构建了一种简单的纤锌矿计算结构及闪锌矿参照结构,分别采用Berry phase方法和最大局域化Wannier函数方法,计算了纤锌矿结构ZnO、AlN及GaN的自发极化和压电系数,计算结果与已经报道的计算值非常接近,发现自发极化与结构参数有着紧密的联系.两种计算方法中,最大局域化Wannier函数方法提供了一种非常直观的观察电子结构的手段,以及定量地研究离子极化和电子极化在自发极化中的贡献,该方法清晰的显示了3种半导体的共价键中具有很强的离子性,这是导致具有自发极化的重要原因之一.
[1]Takan hashi K,Yoshikawa A,Sandhu A.Wide Bandgap Semiconductors:Fundamental Properties and Modern Photonic and Electronic Devices[M].Berlin:Springer,2007:1-24.
[2]Wang Z L,Song J H.Piezoelectric Nanogenerators Based on Zinc Oxide Nanowire Arrays[J].Science,2006,312(5771):242-246.
[3]Makino T,Segawa Y,Tsukazaki A,et al.Photoexcitation screening of the built-in electric field in ZnO single quantum wells[J].Applied Physics Letter,2008,93(12):121907-121909.
[4]Bretagnon T,Lefebvre P,Guillet T,et al.Barrier composition dependence of the internal electric field in ZnO/Zn1xMgxO quantum wells[J].Applied Physics Letter,2007,90(20):201912-201914.
[5]Allen M W,Miller P,Reeves R J,et al.Influence of spontaneous polarization on the electrical and optical properties of bulk,single crystal ZnO[J].Applied Physics Letter,2007,90(6):062104-062106.
[6]陆栋,蒋平.固体物理学[M].北京:高等教育出版社,2011:221-222.
[7]King-Smith R D,Vanderbilt D.Theory of polarization of crystalline solids[J].Physical Review B.1993,47(3):1651-1654.
[8]Marzari N,Vanderbilt D.Maximally localized generalized Wannier functions for composite energy bands[J].Physical Review B,1997,56(20):12847-12865.
[9]Vanderbilt D.Berry-phase theory of proper piezoelectric response[J].Journal of Physics and Chemistry of Solids,2000,61(2):147-151.
[10]Dai S,Dunn M L,Park H S.Piezoelectric constants for ZnO calculated using classical polarizable core shell potentials[J].Nanotechnology,2010,21(44):1-8.
[11]Giannozzi P,Baroni S,Bonini N,et al.QUANTUM ESPRESSO:a modular and open-source software project for quantum simulations of materials[J].Journal of Physics,2009,21(39):1-19.
[12]Mostofi A A,Yates J R,Lee Y S,et al.Wannier90:A Tool for Obtaining Maximally-Localized Wannier Functions[J].Computer Physics Communications,2008,178(9):685-699.
[13]Lhnemann J,Brandt O,Jahn U,et al.Direct experimental determination of the spontaneous polarization of GaN[J].Physical Review B,2012,86(8):0813021(1-5).
[14]Gopal P,Spaldin N A.Polarization,Piezoelectric Constants,and Elastic Constants of ZnO,MgO,and CdO[J].Journal of Electronic Materials,2006,35(4):538-542.
[15]Bernardini F,Fiorentini V.Spontaneous polarization and piezoelectric constants of III-V nitrides[J].Physical Review B,1997,56(16):R10024-R10027.
[16]Farsakh H A,Qteish A.Ionicity scale based on the centers of maximally localized Wannier functions[J].Physical Review B,2007,75(8):085201(1-6).
