壳聚糖/PEI凝胶模板法制备Au/SiO2复合纳米粒子
2014-12-31艾媛媛郑行望
党 婕,艾媛媛,郑行望
(陕西师范大学 化学化工学院,陕西 西安 710062)
近年来,金属等纳米粒子表面包覆二氧化硅所形成核壳结构的复合纳米材料备受关注,这类复合纳米材料因其独特的化学、物理、光学、电学性质以及良好的生物相容性,在催化、材料科学、生物化学传感等领域获得广泛应用[1-8].目前,对金纳米粒子为核和二氧化硅为壳的核壳材料有较多研究,通过对裸金纳米粒子包覆二氧化硅,从而提高金纳米粒子的稳定性和分散性,为开发纳米材料的潜在应用提供良好的契机,展现出很多新的光学、热学、电子学和催化特性[2,9-10].这是因为二氧化硅具有良好的亲水性、生物相容性和光学透明性等特点,通过溶胶-凝胶化学过程易于控制其厚度等优点,从而更好的调节金属纳米粒子的特性[11-13].而且,二氧化硅还可以提高纳米粒子的稳定性并增强其在各种溶液中的溶解度[14].文献中报道的二氧化硅包覆过程,一般通过粒子表面的化学作用力或静电亲和力与二氧化硅作用[15-19].Stober方法是一种最常见的二氧化硅包覆纳米粒子方法.如Mulvaney等人利用硅烷偶联试剂作为引物与金表面结合;然后,加入活性硅酸钠;再通过Stober方法进一步增加硅壳[2].由于其涉及过程复杂,因此不易控制.而Graf等人利用聚乙烯基吡咯烷酮修饰表面后包覆二氧化硅[20].此方法简单快速,但需选择合适的聚合物,因为聚合物的链长短对其均匀性有很大的影响[14,20].因此,在纳米尺度上合成均匀和可控的硅壳厚度仍充满挑战.为了克服此问题,有文献报道将反相微乳液法用于制备二氧化硅包覆金钠米粒子.如Jackie小组采用简单的反相微乳液法制备单分散的二氧化硅包覆金钠米粒子[21].该方法能直接对金纳米粒子和银纳米粒子进行二氧化硅的包覆,没有使用硅烷偶联试剂和聚合物作稳定剂,且对二氧化硅壳的厚度能够比较精准的控制.然而在多数情况下,二氧化硅只包覆一个金纳米粒子并且得到的二氧化硅包覆层厚度不均一,因此,制备连续、光滑、完整的二氧化硅包覆多个金纳米粒子仍然是一个挑战,故本文提出以高分子水凝胶材料为模板,结合反相微乳液合成纳米粒子的技术路线,通过高分子水凝胶模板调节并控制复合纳米材料中金纳米粒子尺寸,为设计合成新型的Au/SiO2复合纳米材料提供了新思路,从而拓宽Au/SiO2复合纳米材料的应用领域.
本文报道我们采用反相微乳液法以壳聚糖/PEI复合水凝胶作为模板制备Au/SiO2核壳结构复合纳米粒子,同时,选择PEI为还原剂直接在反相微乳液中合成金纳米粒子,成功地合成了核壳结构Au/SiO2复合纳米粒子.通过控制复合水凝胶模板中PEI的量,从而使得复合粒子中金粒子尺寸可调.最后,将制备的核壳结构Au/SiO2复合纳米粒子催化硼氢化钠还原4-硝基苯酚(4-NP)的催化活性进行了研究.
1 实验
1.1 试剂和仪器
所用试剂均未作进一步纯化,实验用水均为二次水.正硅酸四乙酯(TEOS)、Triton X-100(TX-100)购于Sigma公司,0.5% 壳聚糖(chitosan分析纯,购于Sigma公司)储备液:准确称取1.000g壳聚糖溶于100mL 1% 醋酸溶液中.聚醚酰亚胺(PEI)、戊二醛购于Sigma公司.其他试剂:正己醇(天津市科密欧化学试剂厂);环己烷、氨水(25%~28%)、丙酮、无水乙醇等均为分析纯(西安化学试剂厂);氯金酸、硼氢化钠和4-硝基苯酚购于国药集团化学试剂有限公司.
多位点磁力搅拌器(德国,IKA)用于纳米粒子合成时的搅拌;5804R型高速冷冻离心机(德国,eppendorf公司)用于纳米粒子的离心分离;JEM-2100型透射电子显微镜(日本,日本电子公司)用于表征纳米粒子结构和粒径;紫外可见吸收光谱用TU-1901双光束紫外可见分光光度计(北京普析通用仪器有限责任公司)检测;原子吸收光谱仪(日本,岛津)用于表征金元素的含量的研究.
1.2 Au/SiO2复合纳米粒子的合成
以壳聚糖/PEI/戊二醛复合水凝胶作为模板,通过反相微乳液法制备核壳型Au/SiO2复合纳米粒子.将环己烷、表面活性剂Triton X-100和助表面活性剂正己醇按体积比4.2∶1∶1混合均匀,加入适量的超纯水混合,搅拌20min后,形成透明状且性质稳定的油包水的微乳液.然后,加入0.5% 壳聚糖溶液搅拌30min,再加入5%的PEI溶液搅拌30 min,使PEI溶液与壳聚糖溶液混合均匀;再加入0.5%氯金酸溶液搅拌30min,随后加入5%的戊二醛溶液,与壳聚糖/PEI复合物交联,生成壳聚糖/PEI/戊二醛刚性复合水凝胶并作为模板,用于生成非连续金纳米粒子.搅拌4h后,将硅烷化试剂TEOS和氨水按1.3∶1的体积比加入到反相微乳液体系中,持续搅拌24h.反应完成后,加入丙酮进行破乳,破乳后纳米颗粒从微乳液体系中析出,离心收集颗粒,依次用无水乙醇、二次水多次洗涤纳米颗粒,以除去表面活性剂分子和助表面活性剂分子,最后将制得的纳米颗粒分散在二次水中并储存4℃的冰箱中备用.
1.3 Au@SiO2核壳型复合纳米粒子的性质表征
1.3.1 复合纳米粒子的大小、形貌表征 利用透射电子显微镜对合成的复合纳米粒子的大小以及形貌进行表征.具体方法为:将合成的复合纳米粒子均匀分散后滴于铜网支撑的碳膜上,充分自然干燥后用透射电子显微镜进行拍照.
1.3.2 不同复合水凝胶为模板所合成的复合纳米粒子中金元素相对含量的表征 利用原子吸收光谱法(AAS)检测这些复合纳米粒子中金的相对含量.将合成的复合纳米粒子放入真空干燥器中干燥后,分别称重;然后,加入0.5mL王水溶解这些复合纳米粒子,再加入9.5mL二次水并在水浴中加热约40min,最终保持体积为10mL,摇匀;取5mL溶液25mL比色管中,加水定容至刻度线,直接上机测量.通过原子吸收光谱的数据分别计算金在复合纳米粒子的相对含量.
2 结果与讨论
2.1 Au@SiO2核壳型复合纳米粒子的合成及表征
2.1.1 壳聚糖/PEI/戊二醛纳米水凝胶的制备壳聚糖加入到反相微乳液后,反相微乳液体系呈现透明且性质稳定.这个结果表明,壳聚糖是一个长链聚合物,但它能稳定的存在于反相微乳液体系中.然而,随后在以上体系中加入PEI,发现反相微乳液体系发生浑浊.其主要原因是由于PEI极好的水溶性能与壳聚糖相混溶,在碱性介质(5%PEI呈强碱性)中形成壳聚糖/PEI水凝胶,使一部分水限制于水凝胶的内部,从而破坏了反相微乳液体系,使反相微乳液体系呈现浑浊.而研究发现将反相微乳液体系中水的总体积从180μL增加到300μL后,PEI的加入可以形成一个性质稳定且透明的一个油包水微乳液体系.接着,将戊二醛加入壳聚糖/PEI复合水凝胶中,发现反相微乳液体系呈现透明且性质稳定.而戊二醛与壳聚糖/PEI复合水凝胶发生交联反应形成具有刚性多孔结构的复合水凝胶,因此,在反相微乳液体系中制备了壳聚糖/PEI/戊二醛复合纳米水凝胶模板.
2.1.2 金纳米粒子在壳聚糖/PEI/戊二醛复合水凝胶中形成 以反相微乳液体系中形成壳聚糖/PEI复合水凝胶基础上,进一步在该复合水凝胶的多孔结构模板中形成金纳米粒子.因此,氯金酸溶液加入反相微乳液体系,并进入已形成壳聚糖/PEI复合水凝胶的多孔结构,之后使加入戊二醛变成刚性复合水凝胶.加入氯金酸溶液,反相微乳液呈微黄色,并透明且性质稳定.4h后,反相微乳液体系呈现红色,并随PEI的量增加,体系呈现的红色由深变浅.同时我们分析了此时各反相微乳液体系的紫外可见光谱,其实验结果如图1所示.由图1可知,527 nm处均有金纳米粒子的表面等离子体共振吸收峰,并随复合水凝胶组分中PEI的量的增大,紫外可见吸收峰值减小.上术实验结果表明:由于PEI上大量—NH2与Au之间强烈的氢键等超分子作用,随着PEI的量增加,使相同量的氯金酸溶液于刚性复合水凝胶材料的多孔结构中生成更多小的金纳米粒子,从而大粒径的金纳米粒子的量减少,因此,527nm处紫外峰值减小.而430nm处的吸收峰的出现可能是因为PEI的—NH2与Au之间相互作用产生的一种新的配合物.
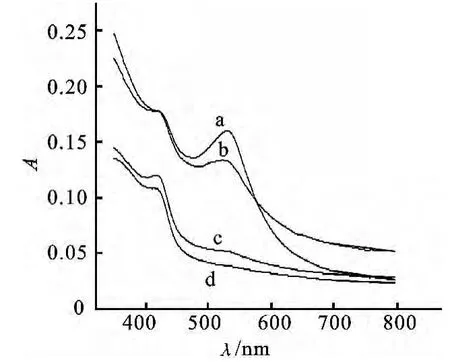
图1 在反相微乳液体系中的Au/复合水凝胶的紫外可见光谱图Fig.1 UV-vis spectra of Au/nanohydrogel in the reverse microemulsion
此外,我们对上述反应中金纳米粒子形成的原因作了研究.在其他实验条件相同的情况下,复合水凝胶制备过程中不加入PEI.研究结果表明:在PEI不存在的情况下,氯金酸加入到反相微乳液体系4 h后未呈现红色并在527nm处没有出现紫外吸收峰.这一现象表明PEI还原氯金酸溶液形成金纳米粒子.因此,我们认为金纳米粒子在复合水凝胶中形成的可能机理为:首先,Au分散于纳米复合水凝胶模板内部的多孔结构,通过Au与PEI上的氨基之间发生配位反应;其次,固定于复合水凝胶的多孔结构的AuCl-4进一步被PEI还原生成金纳米粒子.
2.1.3 核壳型Au/SiO2复合纳米粒子的形成 为了获得核壳结构的多个、不连续Au/SiO2复合纳米粒子,TEOS和氨水加入以上已制备的含有多个金纳米粒子的复合水凝胶,进行二氧化硅包覆.为了考察复合纳米粒子的粒径及形貌,我们采用透射电子显微镜对其形貌进行了表征,图2为复合纳米粒子的TEM照片.由图2a知,Au/SiO2复合纳米粒子是核壳型结构.由图2b可知,复合纳米粒子粒径大约为70nm左右,单分散性较好,分别以含有多个金纳米粒子的复合纳米水凝胶和二氧化硅作为核和壳.位于核部分的复合水凝胶中含有单个或多个的不连续金纳米粒子,其颜色较黑,粒径约为2~5nm左右.

图2 核壳型Au/SiO2复合纳米粒子的TEM图Fig.2 TEM images of the Au@SiO2nanocomposites
2.1.4 PEI量对复合纳米粒子的结构的影响 在成功制备出核壳型Au/SiO2复合纳米粒子的基础上,我们考察了随PEI量的增加,核壳型Au/SiO2复合纳米粒子结构的变化.如图3a—d所示,依次为PEI的用量分别为25、50、75和100μL时最终合成的Au/SiO2复合纳米粒子的TEM图,发现核部分依次生成更小的金纳米粒子.并测定该四种Au/SiO2复合纳米粒子的水溶液对应的紫外可见光谱(如图4),由图可知,该四种Au/SiO2复合纳米粒子的水溶液均在527nm处有紫外吸收峰,说明金纳米粒子被包覆于二氧化硅.另外,由该四种Au/SiO2复合纳米粒子的水溶液的光学照片可知,发现随PEI的量增加,这4种Au/SiO2复合纳米粒子的水溶液的红色由深变浅,与之前在反相微乳液合成过程中,未进行二氧化硅包覆的结果是一致.然而,在PEI量大时,我们在图3d中几乎不能在核部分的复合水凝胶中发现金纳米粒子,但是复合纳米粒子的颜色依然呈红色.这一系列的研究结果可以推测:随PEI的量增加,复合水凝胶中形成更小的金纳米粒子.

图3 不同PEI量的核壳型Au/SiO2复合纳米粒子的TEM图Fig.3 TEM images of Au/SiO2nanocomposites prepared by using the different amount of PEI

图4 不同PEI量的核壳型Au/SiO2复合纳米粒子的紫外光谱图Fig.4 UV-vis spectra of the Au/SiO2nanocomposites prepared by using different amount of PEI
为了验证加入量大的PEI所制备的复合纳米粒子的核部分的复合水凝胶中确实是形成了更小的金纳米粒子,我们采用AAS对上述制备的4种Au/SiO2复合纳米粒子所含金元素相对含量进行考察,实验测定结果如表1.其结果显示,不同PEI量(分别为25、50、75和100μL)所合成的Au/SiO2复合纳米粒子含金元素相对含量几乎是相同的.虽然TEM图中,当PEI量大时生成的复合纳米粒子的核部分几乎看不到有金纳米粒子位于复合水凝胶中,但AAS的结果证明在核部分确实生成了的金纳米粒子,可能是太小,无法分辨.这一系列的结果说明与之前所推测的一样,PEI的量增加,确实是在核部分的复合水凝胶中形成了多个、非连续、更小的金纳米粒子,且在反相微乳液体系中的Au/复合水凝胶和最终制备的Au/SiO2复合纳米粒子水溶液均呈红色,形成的金纳米粒子越小,颜色越浅.因此,所有的结果表明,可以通过改变PEI的用量来调节复合纳米粒子核内的金粒子的大小.

表1 原子吸收光谱的数据Tab.1 Date of atomic absorption spectrometry for different samples
2.2 核壳型Au/SiO2复合纳米粒子催化硼氢化钠还原4-硝基苯酚的催化活性
为了初步研究复合纳米粒子催化硼氢化钠还原4-硝基苯酚反应,我们选取所合成的这些纳米粒子中的一种(25μL PEI合成的核壳型Au/SiO2复合纳米粒子),称取0.314g,用 NaOH (1mL,0.02 mol/L)刻蚀10min,待用.4-硝基苯酚溶液(0.03 mL,0.01mol/L)和硼氢化钠溶液 (0.20mL,0.1 mol/L)加入2.5mL二次水中,并在磁力搅拌器上搅拌.图5a展示了4-NP溶液的紫外吸收峰在317 nm,观察到400nm处强的吸收峰来源于硼氢化钠加入后生成了4-硝基苯酚负离子.通常在无催化剂存在时,即使过量的硼氢化钠也无法促使4-NP转变为4-氨基苯酚.然后加入合成好的复合纳米粒子,可以看到,明亮的黄色溶液逐渐褪色.2h后,溶液的颜色变为无色.同时,测其催化过程的紫外可见光谱图(如图5b),400nm的吸收值减小,这一现象表明4-NP被还原为4-氨基苯酚[22].这从而说明,所合成的纳米粒子有催化作用,催化活性可能与我们合成的复合纳米粒子中核部分的金纳米粒子的尺寸的大小有关,我们接下来将会进一步的研究.
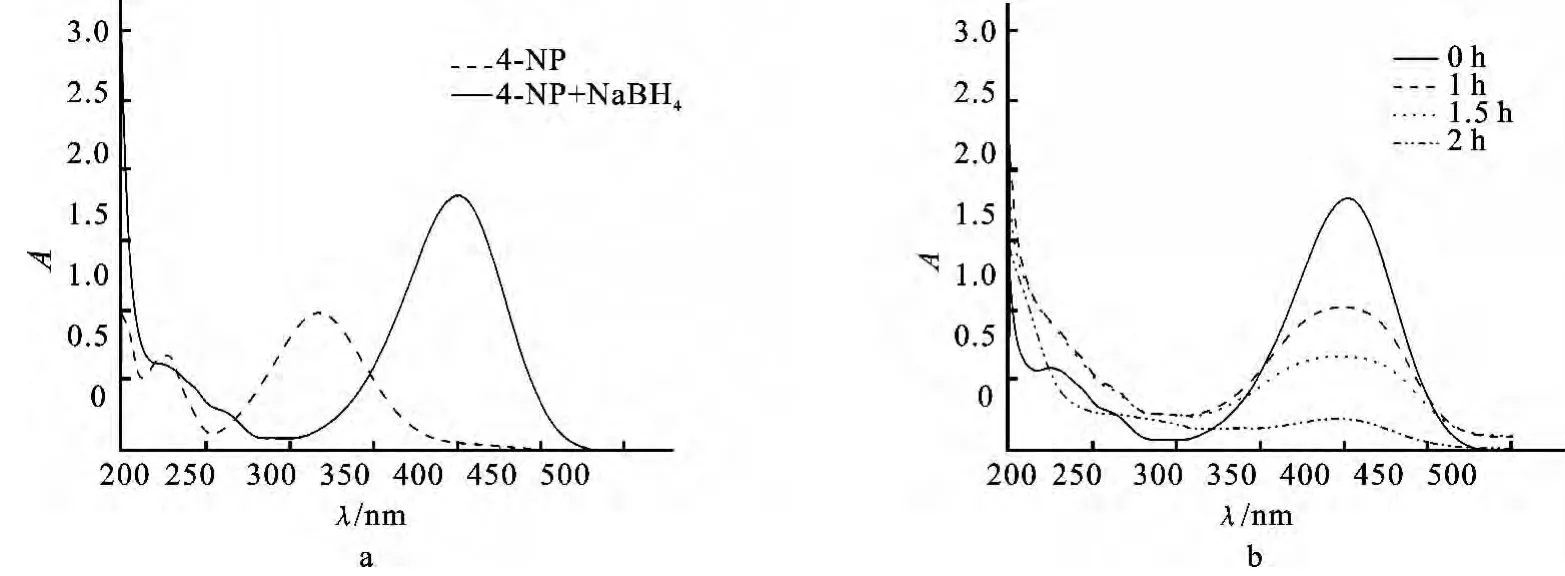
图5 4-NP中加入NaBH4前后的紫外光谱图(a)和加入核壳型Au/SiO2复合纳米粒子催化硼氢化钠还原4-硝基苯酚的紫外光谱图(b)Fig.5 UV-vis absorption spectra of 4-NP before and after addition of NaBH4(a)and addition of NaBH4in presence of Au/SiO2nanocomposites catalyst(b)
3 结论
通过以复合水凝胶作为模板,提出一种新的方法并成功制备了核壳型Au/SiO2复合纳米粒子.这一合成方法的优点是,改变核部分的复合水凝胶中组分的量可以控制复合纳米粒子中金纳米粒子的大小,使其形成多个、非连续、更小的金纳米粒子.与之前报道过的单个的Au/SiO2核壳型纳米粒子相比,这种方法由于合成不连续的金纳米粒子具有较大的比表面积,可以产生新的潜在应用.而且,基于复合水凝胶模板的多功能性质,这种合成方法也可以应用在合成其他金属的核壳型复合纳米粒子.
[1]Graf C,Vossen D L J,Imhof A,et al.A.A general method to coat colloidal particles with silica[J].Langmuir,2003,19:6693-6700.
[2]Liz Marzan L M,Giersig M.Synthesis of nanosized gold-silica core-shell particles[J].Langmuir,1996,12:4329-4335.
[3]Gerion D,Pinaud F,Williams S C,et al.Alivisatos A P.Synthesis and properties of biocompatible water-solu-ble silica-coated CdSe/ZnS semiconductor quantum dots[J].The Journal of Physical Chemistry B,2001,105:8861-8871.
[4]Nann T,Mulvaney P.Single quantum dots in spherical silica particles[J].Angewandte Chemie International E-dition,2004,43:5393-5396.
[5]Santra S,Tapec R,Theodoropoulou N,et al.Synthesis and characterization of silica-coated iron oxide nanoparticles in microemulsion:The effect of nonionic surfactants[J].Langmuir,2001,17:2900-2906.
[6]Philipse A P,Vanbruggen M P B.Magnetic silica dispersions:preparation and stability of surface-modified silica particles with a magnetic core[J].Langmuir,1994,10:92-99.
[7]Mulvaney P,Liz-Marzan L M,Giersig M,et al.Silica encapsulation of quantum dots and metal clusters[J].Journal of Materials Chemistry,2000,10:1259-1270.
[8]Cushing B L,Kolesnichenko V L,O'Connor C.Recent advances in the liquid-phase syntheses of inorganic nanoparticles[J].Chemical Reviews,2004,104:3893-3946.
[9]Lu Yu,Yin Yadong,Mayers B T,et al.Modifying the surface properties of superparamagnetic iron oxide nanoparticles through a sol-gel approach[J].Nano Letters,2002,2(3):183-186.
[10]Zhang Qiao,Zhang Tierui,Ge Jianping,et al.Permeable silica shell through surface-protected etching[J].Nano Letters,2008,8(9):2867-2871.
[11]Salgueirino-Maceira V,Caruso F,Liz-Marzan L M.Coated colloids with tailored optical properties[J].The Journal of Physical Chemistry B, 2003, 107:10990-10994.
[12]Jim nez E,Abderrafi K,Abargues R,et al.Laser-ablation-induced synthesis of SiO2-capped noble metal nanoparticles in a single step[J].Langmuir,2010,26:7458-7463.
[13]Pierre M C,Haes A J.Purification implications on SERS activity of silica coated gold nanospheres[J].Analytical Chemistry,2012,84:7906-7911.
[14]Pastoriza-Santos I,Perez-Juste J,Liz-Marzn L M.Silica-coating and hydrophobation of CTAB-stabilized gold nanorods[J].Chemistry of Materials,2006,18:2465-2467.
[15]Ralph K Iler,Wilmington Dei.Product comprising a skin of dense,hydrated amorphous silica bound upon a core of another soild moterial and process of making same:USA,2885366[P].1959-05-05.
[16]Ohmori M,Matijevic E J.Preparation and properties of uniform coated colloidal particles.Ⅶ.Silica on hem-atite[J].Colloid and Interface Science,1992,150:594-598.
[17]Philipse A P,Van Bruggen M P B.Magnetic silica dispersions:preparation and stability of surface-modified silica particles with a magnetic core[J].Langmuir,1994,10:92-99.
[18]Philipse A P,Nechifor A M.Isotropic and birefringent dispersions of surface modified silica rods with a boehmite-needle core [J]. Langmuir,1994,10:4451-4458.
[19]Chang Songyuan,Liu Lei,Asher S A.Preparation and properties of tailored morphology,monodisperse colloidal silica-cadmium sulfide nanocomposites[J].Journal of the American Chemical Society,1994,116:6739-6744.
[20]Graf C,Vossen D L J,Imhof A,et al.A general method to coat colloidal particles with silica[J].Langmuir,2003,19:6693-6700.
[21]Han Y,Jiang J,Lee S S,et al.Reverse microemulsion-mediated synthesis of silica-coated gold and silver nanoparticles[J].Langmuir,2008,24:5842-5848.
[22]Preeti D,Mausumi M.Prunus domestica fruit extractmediated synthesis of gold nanoparticles and its catalytic activity for 4-nitrophenol reduction[J].Industrial &Engineering Chemistry Research,2012,51:13014–13020.
