黑曲霉DL08内切葡聚糖酶基因的克隆表达及酶学性质研究*
2014-12-16王晓辉张庆芳迟乃玉
王晓辉,张庆芳,迟乃玉
(大连大学生命科学与技术学院,辽宁大连,116622)
纤维素是公认的地球上分布最广、含量最丰富的可再生资源,利用廉价的可再生木质纤维素可转化成葡萄糖,并进一步生成可再生生物能源如乙醇或其他有用物质等。工业生产中主要采用浓硫酸等将长链多聚物纤维素降解为单糖,但所用强酸造成环境严重污染,因此亟需高效率、无污染的纤维素酶替代化学方法。但目前纤维素酶催化效率较低、生产成本较高,影响了纤维素酶工业化生产和广泛应用[1]。
纤维素酶是能够降解纤维素生成葡萄糖的一组酶的总称。主要分为3类:内切葡聚糖酶(endo-1,4-β-D-glucanase,EC3.2.1.4,EG)、外切葡聚糖酶或纤维二糖水解酶(exo-1,4-β-D-glucanases,EC3.2.1.9,CBH) 和 β-葡 萄 糖 苷 酶 (β-glucosidases,EC3.2.1.21,BG)[2-3]。纤维素酶水解纤维素是纤维素酶3种组分协同作用的结果:EG负责进攻纤维素的非结晶区,随机水解β-1,4-糖苷建,将长链纤维素分子截短,产生大量带非还原性末端的小分子纤维素;CBH负责从纤维素线状分子非还原性末端水解切下纤维二糖单位;BG则将纤维二糖水解为葡萄糖,使纤维素完全降解。由于纤维素多样性及高度复杂性,纤维素酶系降解机理还有待进一步研究[4-5]。
为了研究纤维素酶之间的协同作用,需要获得各种纯的纤维素酶制剂。已报道从黑曲霉真菌培养液中分离纯化天然纤维素酶[6-8],但常常存在其他微量多糖水解酶的干扰,最好的办法是利用基因工程菌表达纤维素酶。很多纤维素酶在真核细胞表达系统如酵母细胞或丝状真菌细胞中成功表达,但丝状真菌细胞存在内源纤维素酶干扰问题,酵母细胞中也发现存在内源果胶酶,影响外源基因表达[9-10],因而探讨纤维素酶在原核细胞中异源表达有重要意义。内切葡聚糖酶是纤维素酶系的重要组成部分,占表达的纤维素酶总量的5%~10%,是最重要的内切酶之一,它具有很高的催化活性,被广泛用于纺织、印刷、染料、造纸和洗涤等工业[11]。本文报道来自黑曲霉(Aspergillus niger)DL08内切葡聚糖酶基因首次在大肠杆菌中异源重组表达,并对其酶学性质进行研究,为实现其与外切葡聚糖酶和β-葡萄糖苷酶基因重组并在一个强启动子调控下完成基因串联表达,最终获得纤维素高效降解工程菌鉴定基础。
1 材料与方法
1.1 菌种、质粒和培养条件
黑曲霉(A.niger)DL08菌株由本实验室分离并保存;大肠埃希氏菌(Escherichia coli)DH5α和BL21(DE3)为本实验室保存,分别为基因克隆和表达宿主菌;质粒 pMDl9-T(TaKaRa)用作克隆载体,pET28a(Novagen)用作表达载体;引物合成和基因测序由生工生物工程(上海)股份有限公司完成。
1.2 内切葡聚糖酶基因的克隆与转化
PDA液体培养基活化黑曲霉菌种,取1 mL接入50 mL诱导培养基中,28℃培养48 h后离心收集菌体。用滤纸吸干水分后,加入液氮研磨至粉末状,按照Takara公司的RNA抽提试剂抽提黑曲霉总RNA,并采用cDNA合成试剂盒得到 cDNA。根据 Jiong等[12]发表的内切葡聚糖酶基因序列(GeneBank AF331518)设计引物,正义引物(5’-GAATTCATGAGGCTTCAGAGCACTCTGCTTCTTG-3’)包含EcoRⅠ限制性酶切位点,反义引物(5’-AAGCTTTCAGCGATATGCCTC CAGGAT-3’)包含HindⅢ限制性酶切位点,使用PrimeSTARTMHS DNA Polymerase(Takara)扩增得到不含信号肽的内切葡聚糖酶基因cDNA序列。将该cDNA序列连接到pMDl9-T载体,构建pMDl9-T-eg1质粒转化至E.coli DH5α,得到阳性转化子。用限制性内切酶EcoRⅠ和HindⅢ将eg1基因片段从pMDl9-T-eg1上切下,连接到pET-28a表达载体上,转化至E.coil BL21(DE3),通过菌落PCR、酶切筛选鉴定阳性克隆子,命名为pET28a-eg1。
1.3 内切葡聚糖酶基因的表达与纯化
将重组E.coil BL21(DE3)接入含卡那的LB培养基中,37℃过夜培养14 h。按1%的接种量接种到含卡那的LB培养基中。当OD600nm达到0.8左右,加入0.2 mmol/L的IPTG,15 ℃,100 r/min低温诱导6 h。诱导表达结束后,4℃,5,000×g,4 ℃离心 5 min,收集菌体,用10 mL,20 mmol/L Tris-HCl,50 mmol/L NaCl(pH 8.0)洗涤菌体3次。最后用10 mL含1 mmol/L DTT,1 mmol/L PMSF,20 mmol/L Tris-HCl和50 mmol/L NaCl(pH8.0)的缓冲液重悬菌体。离心管置于冰上超声(400 W)破碎3次,每次30 s,每次间隔1min。4℃,8 000×g离心20 min,收集上清液,即为粗蛋白。粗蛋白用Ni2+-NTA柱(Novagen)进行亲和层析纯化,洗脱的蛋白液转至透析袋(25 mm×16 mm,Sigma-Aldrich)在50 mmol/L Tris-HCl(pH 5.0)缓冲液中4℃过夜透析。蛋白样品经变性聚丙烯酰胺凝胶电泳(SDS-PAGE)[13]检测,其中浓缩胶浓度5%,分离胶浓度12%,用考马斯亮蓝R-250染色。低分子量蛋白 Marker(TakaRa)用于 SDSPAGE。
1.4 蛋白质浓度测定
蛋白浓度通过Bradford蛋白浓度测定试剂盒(Bradford Protein Assay Kit)(碧云天公司)测定。
1.5 内切葡聚糖酶酶学性质测定
1.5.1 内切葡聚糖酶的酶活测定
内切葡聚糖酶活性采用3,5-二硝基水杨酸试剂(DNS)法[14]测定。反应体系为:0.3 mL,50 mmol/L Tris-HCl buffer(pH8.5),90 μL,1% 羧甲基纤维素钠和适量重组内切葡聚糖酶,45℃反应30 min。加0.8 mL,0.4 mmol/L DNS试剂终止反应,100 ℃ 加热 10 min显色,然后检测在520 nm处的吸收值,根据标准曲线回归方程,得到还原糖浓度。酶活力单位(U)定义为:在上述条件下每分钟催化底物生成1 μmol还原糖所需酶量为1个活力单位。
1.5.2 内切葡聚糖酶反应的最适温度和温度稳定性
将加入羧甲基纤维素钠的酶液在30、35、40、45、50、55、60、65、70、75、80 ℃下反应 30 min 测酶活,确定酶反应的最适温度。将最高酶活定义为100%。热稳定性的测定是将酶液分别置于30、35、40、45、50、55、60、65、70、75、80 ℃下保温1 h 测酶活,将最高酶活定义为100%。
1.5.3 内切葡聚糖酶反应的最适pH和pH稳定性
最适pH测定所用缓冲液为:50 mmol/L phosphate-citrate(pH 4.0~5.0),50 mmol/L Tris – HCl(pH 6.0~10.0),反应体系为:90 μL,1% 羧甲基纤维素钠,10 μL 酶液,300 μL 不同 pH 的缓冲液,45 ℃反应30 min测酶活,将最大酶活设为100%。pH稳定性的测定:将酶保存于不同pH值的缓冲液中,于4℃放置24 h后在最适pH值和最适温度下测定酶活,以酶活最高为100%。
1.6 酶作用产物分析
硅胶薄层层析(TLC)法分析酶解产物。水解条件:1%羧甲基纤维素钠,0.1 mg重组内切葡聚糖酶和0.01%(w/v)NaN3,45 ℃反应,分别于 4,8,12 和24 h取样。各取10 μL点样于薄层层析硅胶板上,用丙酮∶异丙醇∶水 =6∶3∶1.5作为展开剂,用吹风机吹干硅胶板,均匀喷洒显色剂(4 g二苯胺,4 mL苯胺与20 mL,85%磷酸溶于200 mL丙酮),180℃烘烤3 min,显色。lmg/mL葡糖糖作为标准液。
2 结果与分析
2.1 内切葡聚糖酶基因的克隆与鉴定
以黑曲霉(A.niger)总RNA为模板,RT-PCR扩增,得到1条长度约1000bp的 DNA片段(图1)。DNA测序结果表明:该基因包含999个核苷酸构成的开放阅读框架,命名为eg1,该基因编码332个氨基酸,GC 碱基含量为53.95%,理论分子量36.75 kDa,等电点为4.38。Blast比对分析该序列同已发表A.niger内切葡聚糖酶 (GeneBank No.AF331518)氨基酸同源性达96%。已将此基因登陆GenBank,序列号为KJ437592。结构域分析[17]该基因包含一段18个氨基酸构成的信号肽,属于糖苷水解酶5家族。将目的基因eg1连接表达载体pET28a,酶切鉴定成功构建表达载体pET28a-eg1(图2)。
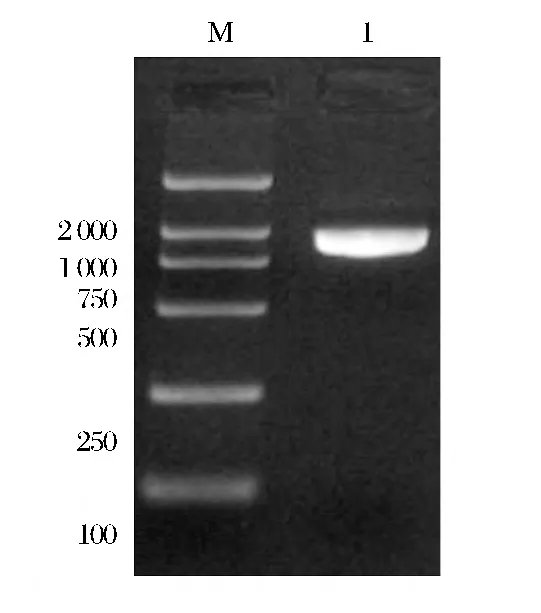
图1 内切葡聚糖酶基因的PCR扩增Fig.1 Agarose gel electrophoresis of the eg1 gene through PCR

图2 重组质粒酶切产物回收Fig.2 Enzyme digestion of plasmid pET28a-eg1
2.2 重组内切葡聚糖酶表达与纯化
重组表达质粒载体pET28a-eg1转化至宿主菌E.coli BL21(DE3),0.2 mmol/L IPTG,100 r/min,15℃低温诱导表达6 h,超声破碎后蛋白5.23 mg,全部以可溶蛋白表达,未见包涵体形式,表明该重组蛋白成功表达(见图3),比活力为68.9 U/mg。利用设计好的内切葡聚糖酶基因C端融合6个组氨酸标签,通过Ni+-NTA亲和层析法纯化分离重组内切葡聚糖酶(见图4)。SDS-PAGE检测显示该重组蛋白纯化后得到单一条带,大小约36 kDa,与预期大小一致。蛋白含量2.85 mg,纯化倍数为1.31,回收率达59.2%,比活力高达 90.3 U/mg(见表 1)。

图3 重组内切葡聚糖酶蛋白SDS-PAGE检测结果Fig.3 Sodium dodecyl sulfate polyacrylamide gel electrophoresis(SDS-PAGE)of the recombinant eg1

表1 内切葡聚糖酶重组蛋白纯化总结Table 1 Purification summary of the recombinant eg1
2.3 重组内切葡聚糖酶酶学性质研究
由图5(a)可以看出,反应温度在40~60℃时该酶有较高的活性,温度低于40℃或者高于60℃,酶促反应速率显著下降,酶的最适反应温度为45℃;30~60℃区间酶的稳定性较好,其相对酶活都大于80%,说明该酶具有较好的热稳定性;由图5(b)可以看出,该酶最适 pH为5.0,pH大于7.0酶的活力有所下降;该酶在3.0~12.0范围内有较好的活性,保持50%以上的酶活性,pH在表明内切葡聚糖酶pH耐受性范围较广。
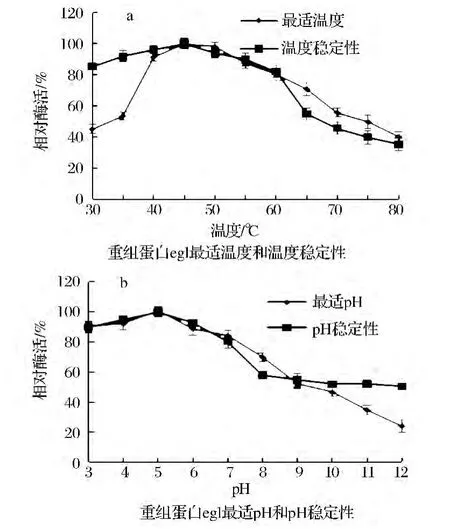
图5 内切葡聚糖酶重组蛋白酶学特性Fig.5 Enzymatic profiles of recombinant endo-β-1,4-glucanase eg1.
2.4 重组内切葡聚糖酶酶解产物分析
纯化的重组蛋白eg1在45℃酶解1%羧甲基纤维素钠,分别于2,4,12,24 h 取 10 μL 溶液进行产物分析。以l mg/mL的葡萄糖为标准液(见图4),从图4看出,该酶的酶解产物为连续的寡糖,而非单一分子质量的单糖产物,说明该酶为内切葡聚糖酶。
3 讨论
异源表达蛋白尤其是来自真核生物的蛋白在大肠杆菌中往往不能正确折叠,而聚集成不溶性的形式即包涵体,造成这些蛋白在大肠杆菌中不能正确表达[15]。研究者采用很多措施改善或提高蛋白的正确折叠效率包括共表达同源或异源的折叠辅助蛋白、菌体培养条件的优化及表达融合蛋白等,其中培养条件的优化是最简单有效促进蛋白正确折叠的方法,包括诱导剂浓度、培养温度或摇床转数等方面,而优化低温诱导是一种常见有效的提高蛋白折叠的策略,其原理可能涉及很多因素,如蛋白自联作用力的减弱、蛋白合成速度的降低以及多肽链折叠动力学的改变等[16]。本研究中内切葡聚糖酶在37℃诱导表达时重组蛋白几乎全部以不溶性的包涵体形式表达,当采用15℃低温诱导表达可溶性的重组蛋白。
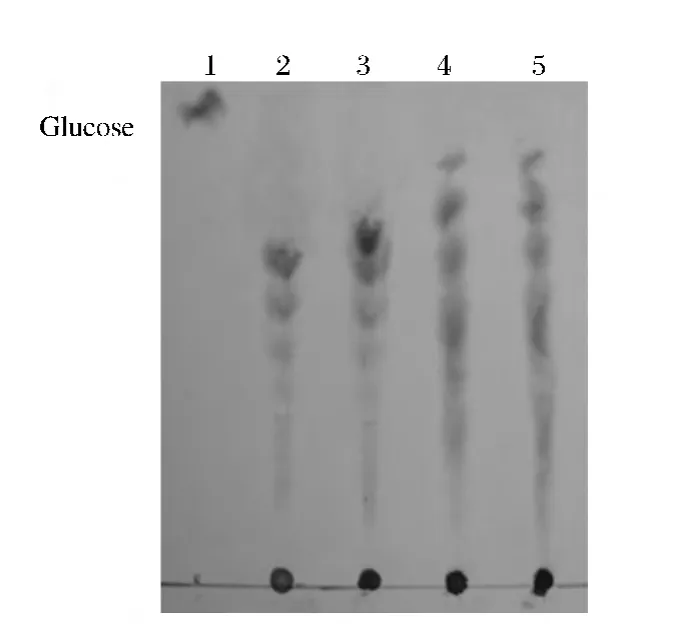
图6 内切葡聚糖酶酶解产物薄层层析Fig.6 TLC analysis of the oligosaccharides from the endo-β-1,4-glucanase hydrolysates
本研究内切葡聚糖酶以羧甲基纤维素钠为底物测酶活,比活力达90.3 U/mg,最适作用温度45℃,为中温酶;最适 pH 为 5.0,在 pH 3.0~12.0 酶活较稳定,即使在强碱性环境下仍保持50%以上的活性。以羧甲基纤维素钠为底物分析水解产物表明4h的酶解产物已产生淡淡斑点,即产生连续低聚寡糖,12和24 h酶解产物相比4 h产生更多斑点,说明随着酶解反应的进行产生浓度更高的低聚寡糖,12 h与24 h酶解产物的斑点并未明显区别,表明该酶于12 h已反应完全,所以24 h的酶解产物在TLC板上并未产生新的斑点,即未产生新的相应低聚寡糖。本研究内切葡聚糖酶酶解1%羧甲基纤维素钠终产物为连续寡糖,并非单一分子质量的单糖,说明该酶为内切葡聚糖酶。
本研究利用诱导型T7启动子控制内切葡聚糖酶基因在大肠杆菌中实现了过量表达,初步研究重组蛋白的酶学特性,并对其降解产物进行定性分析,目前正在对该酶的酶学性质作更深入的研究,为实现其与外切葡聚糖酶和β-葡萄糖苷酶基因重组并在一个强启动子调控下完成基因串联表达,最终获得纤维素高效降解工程菌鉴定基础。
[1]Vaaje-Kolstad,G,Westereng B,Horn SJ,et al.An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides[J].Science,2010,330(6001):219-222.
[2]Westereng B,Ishida T,Vaaje-Kolstad G,et al.The putative endoglucanase PcGH61D from Phanerochaete chrysosporium is a metal-dependent oxidative enzyme that cleaves cellulose[J].PLoS ONE,2011,6(11):1-11.
[3]LI Yanhong,YIN Qiuyu,DING Ming,et al.Purification,characterization and molecular cloning of a novel endo-β-1,4-glucanase AC-EG65 from the mollusc Ampullari acrossean[J].Comparative Biochemistry and Physiology Part B:Biochemistry and Molecular Biology,2009,153(2):149-156.
[4]SW Kang,YS Park,JS Lee,et al.Production of cellulases and hemicellulases by Aspergillus niger KK2 from lignocellulosic biomass[J].Bioresource Technology,2004,91(2):153-156.
[5]Alinda A Hasper,Ester Dekkers,Marc van Mil,et al.EglC,a New Endo-β-1,4-glucanase from Aspergillus niger with major activity towards xyloglucan[J].Applied and Environmental Microbiology,2002,68(4):1 556-1 560.
[6]HUANG Xiaomei,FAN Jinxia,YANG Qian,et al.Cloning,Expression,and characterization of endo-β-1,4-glucanase gene egIV from Trichoderma viride AS 3.3711[J].Journal of Microbiology and Biotechnology,2012,22(3):390-399.
[7]LIN Ling,MENG Xin,FU Peng,et al.Improved catalytic efficiency of endo-β-1,4-glucanase from Bacillus subtilis BME-15 by direct evolution[J].Applied and Environmental Microbiology,2009,82:671-679.
[8]Dominic DW,WS Wong,Victor J Chan,et al.A novel xyloglucan-specific endo-β-1, 4-glucanase:biochemical properties and inhibition studies[J].Applied and Environmental Microbiology,2010,86:1 463-1 471.
[9]QIN Yuqi,WEI Xiaomin,LIU Xiangmei,et al.Purification and characterization of recombinant endo-β-1,4-glucanase of Trichoderma reesei expressed in Saccharomyces cerevisiae with higher glycosylation and stability[J].Protein Expression and Purification,2008,58(1):162-167.
[10]LIU J,LIU W,ZHAO X,et al.Cloning and functional characterization of a novel endo-β-1,4-glucanase gene from a soil-derived metagenomic library[J].Applied Microbiology and Biotechnology,2011,89(4):1 083-1 092.
[11]Svein Jarle Horn,Gustav Vaaje-Kolstad,Bj?rge Westereng,et al.Novel enzymes for the degradation of cellulose[J].Biotechnology for Biofuelsa,2012,5(45):1-12.
[12]Jiong hong,hisanori Tamaki,Shunichi Akiba,et al.Cloning of a gene encoding a highly stable endo-β-1,4-glucanase from Aspergillus niger and its expression in yeast[J].Journal of Bioscience and Bioengineering,2001,92(5):434-441.
[13]Laemmli,U.K.Cleavage of structural proteins during the assembly of the head of bacteriophage T4[J].Nature,1970,227(5259):680-685.
[14]Miller GL.Use of dinitrosalicylic acid reagent for determination of reducing sugar[J].Analytical Chemistry,1959,31:426–428.
[15]Schultz J,Copley RR,Doerks T,et al.SMART:a webbased tool for the study of genetically mobile domains[J].Nucleic acids research,2000,28(1):231-234.
[16]Harming G,Makrides S C.Strategies for optimizing heterologous protein expression in Escherichia coli[J].Trends in Biotechnology,1998,16(2):54-60.
