小分子蛋白酪氨酸激酶抑制剂的合成研究进展
2014-10-11赵迎春张前军卢永仲陈海燕
赵迎春,张前军*,卢永仲,2,陈海燕
(1.贵州大学 化学与化工学院,贵州 贵阳550025; 2.贵州理工学院 药学院,贵州 贵阳550007)
蛋白酪氨酸激酶(PTK)在正常细胞分裂和异常细胞增殖中起着关键作用,能催化ATP的磷酸基,使其转移到许多重要蛋白质的酪氨酸残基上,使酚羟基磷酸化[1].但是在一般的正常细胞中(神经细胞除外),蛋白质的酪氨酸磷酸化是极少发生的.在带有酪氨酸激酶活性的癌基因病毒转化的细胞中,蛋白质上磷酸化的酪氨酸增加了10倍,因此蛋白质酪氨酸激酶的磷酸化作用是细胞转化和恶性增长机制的一部分,他们的异常表达将导致细胞增殖发生紊乱,进而导致肿瘤的形成[2].超过50%的原癌基因和癌基因产物都具有蛋白酪氨酸激酶活性,因此以蛋白酪氨酸激酶PTK作为肿瘤治疗靶点的研究尤其受到关注.目前主要有两种途径可以终止酪氨酸激酶所介导的增殖信号,一类是单克隆抗体,另一类是小分子药物.小分子药物通过与胞内酪氨酸激酶催化区结合,抑制其催化活性,从而阻断细胞增殖信号[3].至今已有10余种PTKs抑制剂和抗体进入I~III期临床试验阶段,个别的已经上市.小分子蛋白酪氨酸激酶抑制剂的分子结构最常见的是喹唑啉、吲哚、喹啉和嘧啶类化合物.本文作者对近十年来含有不同结构类型的蛋白酪氨酸激酶抑制剂的合成及活性研究进展进行综述,为酪氨酸激酶抑制剂的研究与开发提供参考.
1 喹唑啉类
喹唑啉类化合物具有抗肿瘤、抗病毒、抗菌等多种药理作用,酪氨酸激酶抑制剂中含喹唑啉结构的化合物抑制PTK活性最高,选择性最好.目前进入临床试验和临床试用阶段的酪氨酸酶抑制剂中含有喹唑啉结构的药物有吉非替尼和埃罗替尼等[4].

图1 吉非替尼和埃罗替尼结构Fig.1 The structure of gefitinib and erlotinib
KLUTCHKO等人[5]以6-氨基喹唑啉或6-氨基吡啶并嘧啶类化合物和炔酸在吡啶中经缩合反应制备了一系列的包含喹唑啉和吡啶并[3,4-d]嘧啶结构的丙炔酰胺衍生物,该类化合物对表皮生长因子受体激酶pan-ErbB具有广泛的抑制作用,其中化合物4、6、8对ErbB1的平均抑制作用是对ErbB2、ErbB4的10倍(见表1).其合成路线和化合物的生物活性如图1和表1所示:
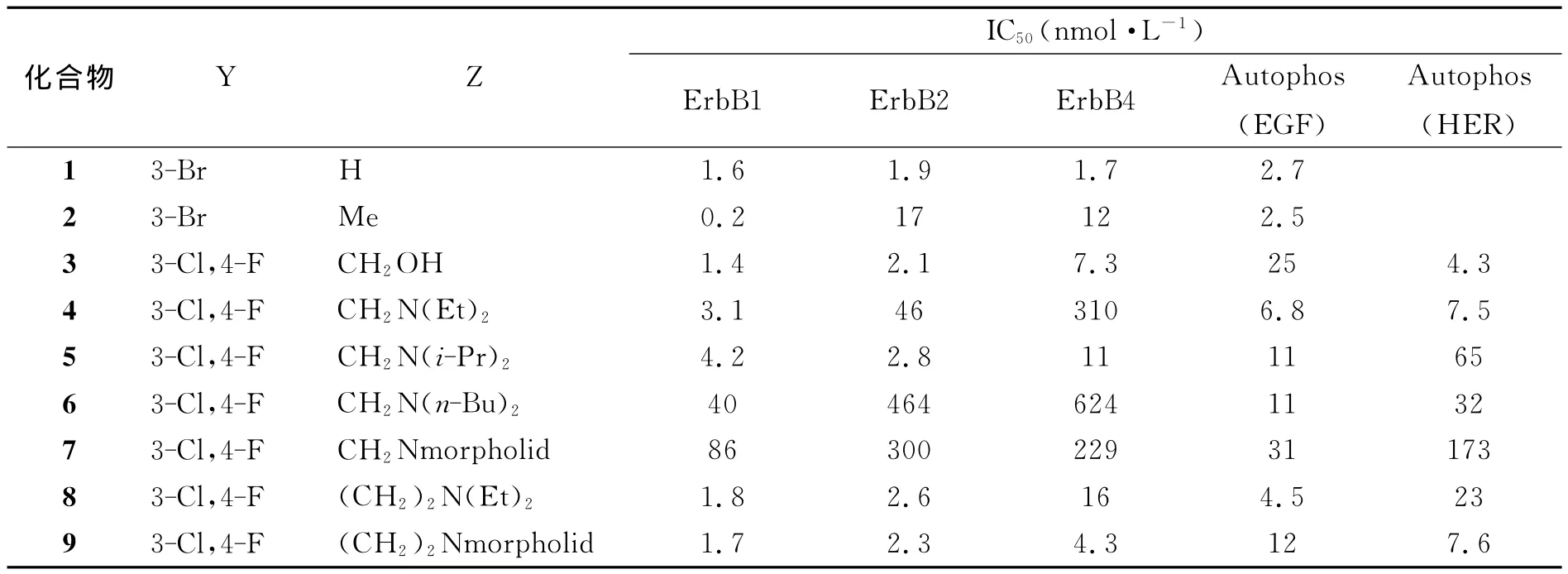
表1 6-氨基喹唑啉类化合物对表皮生长因子受体的抑制活性Table 1 Kinase Inhibitory Properties of 6-aminoquinazoline
C-Kit是一种具有致瘤性的跨膜酪氨酸激酶蛋白,是酪氨酸激酶受体 (TKRs)的成员之一.C-Kit的抑制剂具有潜在治疗肥大细胞相关的纤维化疾病的能力.HU[6]等人报道了25个芳氨基喹唑啉吡啶酮类化合物作为C-Kit的强效抑制剂,抑制作用是血管内表皮生长因子受体(KDR)、丝裂原活化蛋白激酶 (p38)、淋巴细胞特异性蛋白酪氨酸激酶(Lck)和蛋白酪氨酸磷酸酶(Src)的200倍,具有理想的药代动力学特性.通过在啮齿类动物肥大细胞活化的药效学模型实验证明了化合物10具有抑制体内依赖性组织胺释放的作用,有望成为治疗肝纤维化疾病的药物,其合成路线如图3所示:

图3 芳氨基喹唑啉吡啶酮合成路线及化合物10的结构Fig.3 The synthetic route of aryl aminoquinazoline pyridones and the structure of compound 10
VANBROCKLIN等人[7]通过表皮生长因子受体(EGFR)酪氨酸激酶放射性实验证明了苯胺二烷氧基喹唑啉化合物对EGFR酪氨酸激酶有一定的亲和性,可作为筛选表皮生长因子受体酪氨酸激酶抑制剂的潜在的肿瘤成像探针(图4).其中化合物4-(2′-氟苯胺基)6,7-二乙氧基喹唑啉(11a)、4-(3′-氟苯胺基)6,7-二乙氧基喹唑啉(11b)、4-(2′-氯苯胺基)6,7-二甲氧基喹唑啉(12a)和4-(3′-溴苯胺基)6,7-二甲氧基喹唑啉(12b)(图5)与EGFR酪氨酸激酶有较强的结合能力.

图4 苯胺二烷氧基喹唑啉化合物的合成路线Fig.4 The synthetic route of anilinodialkoxyquinazolines
BANERJEE等人[8]设计合成了含喹唑啉的叠氮化合物(图6),对表皮生长因子受体酪氨酸激酶有明显抑制作用,通过单细胞微电泳检测试验证明这些化合物具有破坏整个癌细胞基因组DNA的能力(表2).

图5 化合物11a-12b结构Fig.5 The structure of compounds 11a-12b
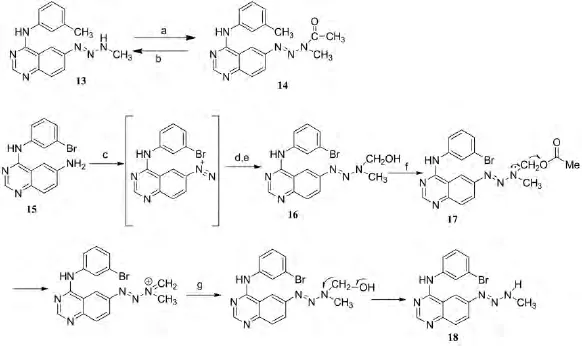
图6 喹唑啉类叠氮化合物的合成路线Fig.6 The synthetic route of acetoxymethyltriazene

表2 喹唑啉类叠氮化合物的EGFR抑制活性Table 2 EGFR TK inhibition of compounds 13-18
2 吲哚类
陈重等人[9]以舒尼替尼为先导化合物,参照已有的构效关系,合成了9个3-取代吲哚-2-酮类目标产物.并以舒尼替尼为阳性对照,采用MTT法测试了目标产物对VEGFR-2高表达的乳腺上皮细胞(HMEC)的抑制活性.样品浓度为10μmol·L-1时,化合物19对VEGFR-2的抑制活性优于阳性对照舒尼替尼.其合成路线如图7所示:
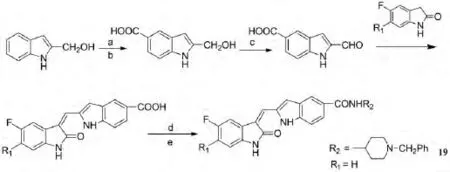
图7 3-取代吲哚-2-酮类化合物的合成路线及化合物19的结构Fig.7 The synthetic route of 3-substituted indolin-2-ones and the structure of compound 19
为了提高抗肿瘤特性和优化药物溶解度和与蛋白的结合度,SUN等[10]研究合成了一系列吲哚-2-酮类衍生物,其中5-[5-Fluoro-2-oxo-1,2-dihydroindol-(3Z)-ylidenemethyl]-2,4-dimethyl-1H-pyrrole-3-carboxylic acid(2-diethylamino-ethyl)amide(化合物20)在生物化学和细胞水平下对血管内皮生长因子(VEGFR2)和血小板衍生的生长因子(PDGF-R)酪氨酸激酶有很好的抑制作用,且具有溶解度高,与蛋白结合能力强及生物利用度好的特点,其半数抑制浓度IC50<0.07μmol·L-1,目前已进入I期临床用于癌症治疗.其合成路线和化合物如图8所示:
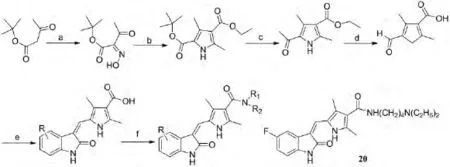
图8 二氢吲哚2-酮类衍生物的合成路线及化合物20的结构Fig.8 The synthetic route of indolin-2-ones devarites and the structure of compound 20
对于急性骨髓性白血病,FMS样的酪氨酸激酶3(FLT3)受体酪氨酸激酶是异常活跃的.MAHBOOBI等人[11]设计合成的化合物21和22能够抑制FLT3和PDGFR激酶,其半数抑制浓度IC50分别为0.4和0.6 μmol·L-1.对PDGFR激酶的选择性高达20~40倍.其合成路线和结构如图9和图10所示:

图9 双(1H-吲哚-2-基)甲基酮类化合物的合成路线Fig.9 The synthetic route of bis(1H-indol-2-yl)methanones

图10 化合物21和22的结构Fig.10 The structure of compounds 21 and 22
3 喹啉类
史祥飞等[12]以2-甲基-2苯基丙酸为原料,经过酯化、硝化等9步反应合成了12个喹啉衍生物,可作为潜在的蛋白酪氨酸激酶抑制剂.其合成路线如图11所示:
KUBO等人[13],发现了 N-苯基-N′-{4-(4-喹啉基氧基)苯基}脲类化合物(结构见图12)不但能够有效抑制VEGFR-2磷酸化,而且对PDGFR家族中的PDGFR和C-Kit也有抑制作用;裸鼠体内实验证明该类化合物对胃癌、肺癌、结肠癌和黑色素瘤等有明显的抗肿瘤活性.

图11 喹啉衍生物的合成路线Fig.11 The synthetic route of quinoline derivatives

图12 N-苯基-N′-{4-(4-喹啉基氧基)苯基}脲类化合物的合成路线Fig.12 The synthetic route of N-phenyl-N′-{4-(4-quinolyloxy)phenyl}ureas
前列腺表皮细胞中存在一种非受体酪氨酸酶,即BMX激酶.BMX激酶表达量过高,导致下游信号途径激活,从而导致前列腺癌细胞转化、增殖等.LIU[14]基于结构的药物设计方法,设计合成了以BMX激酶为靶点的小分子化合物BMX-IN-1(化合物23),对重组BMX激酶具有很好的抑制活性(IC50=8nmol·L-1),能够有效抑制 TEL-BMX转化Ba/F3细胞的增殖(IC50=25nmol·L-1).BMX-IN-1是目前为止第一个被报道的高选择性BMX激酶抑制剂,它的发现为今后开发以BMX激酶为靶点的抗癌症药物奠定了基础.

图13 化合物23的结构Fig.13 The structure of compound 23
4 嘧啶类
FREY等人[15]设计合成了一系列7-氨基吡唑并[1,5-a]嘧啶类化合物,具有强效抑制血管内皮生长因子受体(VEGFR)和血小板衍生生长因子受体(PDGFR)激酶生长的能力,同时对雌激素-诱导的小鼠子宫水肿模型有明显的药代动力学特征.其合成路线、结构以及生物活性见图14和表3:

图14 7-氨基吡唑并[1,5-a]嘧啶类衍生物的合成路线Fig.14 The synthetic route of 7-aminopyrazolo[1,5-a]pyrimidines
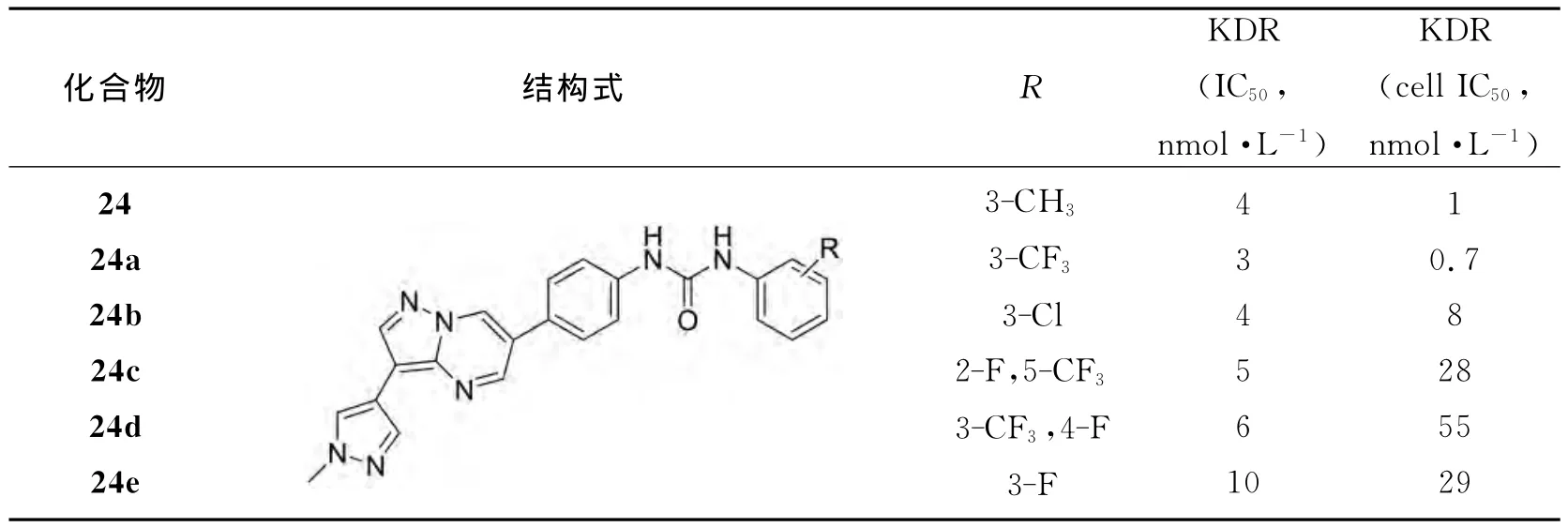
表3 4-甲基吡唑取代吡咯并[1,5-a]嘧啶脲对KDR酶和细胞的抑制活性以及在体内的子宫水肿的抑制活性Table 3 KDR enzymatic and cellular inhibitory activity and in vivo oral uterine edema inhibitory activity of 4-methylpyrazole-substituted pyrazolo[1,5-a]pyrimidine ureas
DAI等人[16]通过构效关系设计合成了一系列以噻吩并嘧啶为母体的化合物,研究了他们对血管内皮生长因子受体和血小板衍生生长因子受体激酶的抑制活性和作用机理,其中化合物25和26具有良好的药代动力学特征,并且表现出很强的抗肿瘤活性.其合成路线和结构分别见图15和图16:

图15 噻吩并嘧啶类化合物的合成路线Fig.15 The synthetic route of thienopyrimidine derivatives

图16 化合物25和26的结构Fig.16 The structure of compounds 25 and 26
KLUTCHKO等人[5]设计合成吡啶并[3,4-d]嘧啶类化合物27(图17),其具有ErbB激酶抑制活性,在体内实验中,对人体表皮样癌A431(用量1mg/kg)、胶质瘤SF767、人卵巢癌SKOV3人胰腺癌BXPC3和人非小细胞肺癌H125(40mg/kg)等癌细胞有很强的抑制作用.

图17 化合物27和28的结构Fig.17 The structure of compound 27 and 28
HURLEY[17]等人报道了苯并呋喃[3,2-d]嘧啶衍生物具有酪氨酸激酶抑制活性.其中化合物28(图17)是多靶点的酪氨酸激酶抑制剂,对GIST882癌细胞株半数抑制浓度为1.6μmol·L-1,对胰腺癌细胞株MIAPaCa和PANC-1的半数抑制浓度分别为2.1和3.0μmol·L-1.该化合物已被SuperGen公司开发为抗癌候选药物并进入一期临床试验阶段.
BRIDGES[18]等人研究发现化合物29具有抑制表皮生长因子受体酪氨酸激酶的作用,对人体表皮癌细胞A431的IC50值为740nmol·L-1.合成路线如图18所示:

图18 化合物29的合成路线Fig.18 The synthetic route of 29
GANGNJEE[19]等设计合成的6种新颖 N4-取代苯基-6-取代苯甲基-7H-吡咯并[2,3-d]嘧啶-2,4-二胺可作为多种受体酪氨酸激酶的抑制剂,其中化合物30、31和32对多种RTK显示出强有效的抑制作用,化合物33和34对VEGFR-2((0.048±0.06)和(0.1±0.021)μmol·L-1)的强效抑制作用明显超过了PDGFR-β((193.2±20.1)和(145±23.8)μmol·L-1)和VEGFR-1(>200和(185.6±27.5)μmol·L-1).其结构见图19:
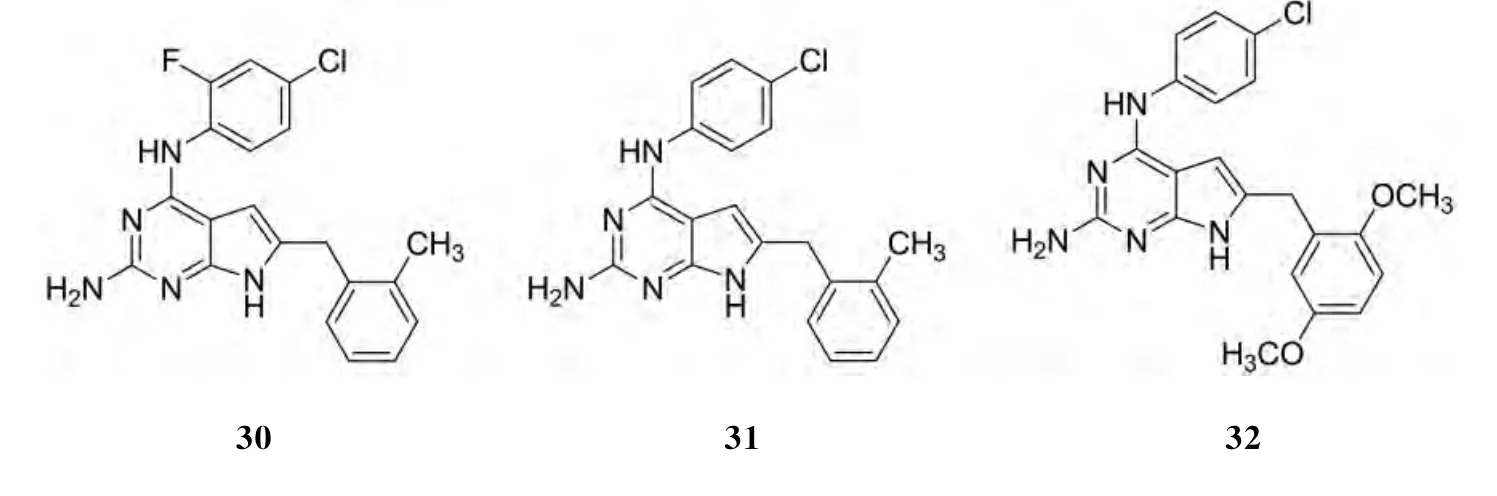
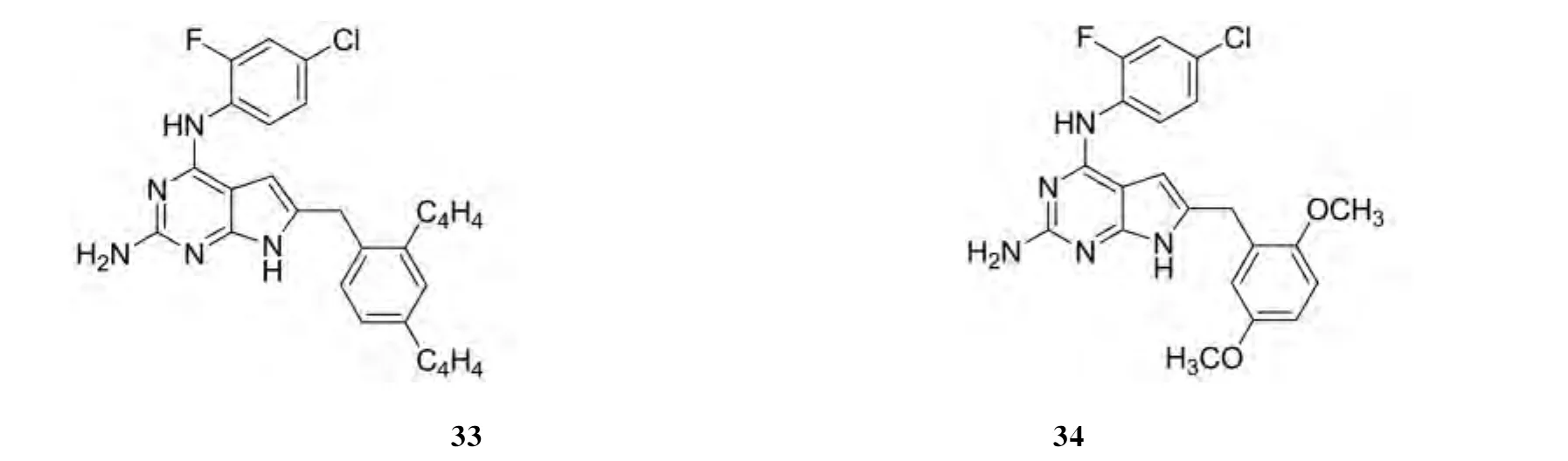
图19 化合物30-34的结构Fig.19 The structure of compounds 30-34
5 其他结构
LANG[20]等设计合成的五个9-苯乙胺基吖啶类化合物35-39可作为潜在酪氨酸激酶和拓扑异构酶I的多靶点抑制剂,对人肝癌细胞HEPC-2显示强抗增殖活性.其中化合物39,对酪氨酸激酶血管内皮生长因子受体(VEGFR-2)和DNA拓扑异构酶(Src)具有抑制活性,其合成路线和活性见图20和表4、表5:
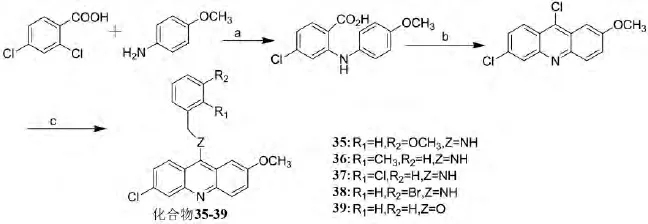
图20 化合物35-39的合成路线Fig.20 The synthetic route of compounds 35-39

表4 化合物35-39对HepG-2细胞的抗增殖活性Table 4 Antiproliferative activity of compounds 35-39 against HepG-2cells

表5 化合物38和39在20μmol/L时对Src和VEGFR-2两种酶的抑制率Table 5 Percent inhibition effect of compounds 38 and 39selected at 20μmol/L on the activity of two kinases
CRAWFORD等人[21]报道了8-氟酞嗪-1(2H)-酮的衍生物(图21)可作为布鲁顿酪氨酸激酶抑制剂,用于治疗各种炎症、免疫疾病和癌症.化合物40、41和42通过CD69Hu血流式细胞仪测定实验分别得到IC70为0.006 0、0.005 9和0.027 2μmol·L-1.

图21 化合物40-42的结构Fig.21 The structure of compounds 40-42
BAINDUR等人[22]合成的三嗪类化合物43(图22)对VEGF-R2(KDR)酪氨酸激酶抑制作用非常强(IC50=18nmol·L-1),同时在人脐静脉血管内皮细胞(HUVEC)的KDR-诱导 MAP激酶的检测中表现出微摩尔级的抑制作用,IC50值为280nmol·L-1.此外通过阻断大鼠主动脉血管内皮环植实验,显示出该化合物有抗血管形成的活性,IC50值为1μmol·L-1.

图22 三嗪类化合物的合成路线和化合物43的结构Fig.22 The synthetic route of triazines and the structure of compound 43
GINGRICH等人[23]报道了以茚并吡咯并咔唑为模板合成的一系列新的强效的血管内皮生长因子R2酪氨酸激酶抑制剂.其中化合物44和45对血管内皮生长因子R2酪氨酸激酶有很强的抑制作用,其IC50值分别为4和8nmol·L-1.化合物46对人类的 VEGF-R1/FLT-1、VEGF-R2/KDR和 VEGF-R3/FLT-4有强效的抑制作用,其IC50值分别为16、8和4nmol·L-1.此外化合物43对酪氨酸和丝氨酸/苏氨酸激酶包括PKC、Tie2、TrkA、CDK1、p38,JNK和IRK等具有良好的选择性,并且在抗肿瘤模型和I期临床试验中,表现出显著的体内抗肿瘤活性(图23).

图23 化合物44-46的结构Fig.23 The structure of compounds 44-46
HO等人[24]设计合成了一系列的(6,7-二甲氧基-1,4-二氢茚并[1,2-c]吡唑-3-基)苯胺类化合物,其中化合物47具有抗PDGFR-激酶的活性(IC50=0.004 2μmol·L-1)和很强的抗肿瘤细胞增殖的活性(IC50<0.033μmol·L-1).合成路线和结构见图24和图25:

图24 (6,7-二甲氧基-1,4-二氢茚并[1,2-c]吡唑-3-基)苯胺类化合物的合成路线Fig.24 The synthetic route of(6,7-dimethoxy-2,4-dihydroindeno[1,2-c]pyrazol-3-yl)phenylamines

图25 化合物47和48结构Fig.25 The structure of compounds 47 and 48
COMEAU等人[25]报道化合物48对蛋白酪氨酸磷酸酯酶1B(PTP1B)的IC50值是220nmol·L-1,并且对PTP1B选择性是对磷酸酶(TCPTP)的30倍.
CHEKLER[26]等设计合成了一类双VEGFR-2激酶和微管蛋白抑制剂.其中化合物49与以往的微管蛋白抑制剂药物相比有很多优点,具有良好的耐受性和疗效性,可以口服给药,可作为治疗癌症的潜在新疗法.
FLT3是一种受体酪氨酸激酶(RTK),是急性髓细胞白血病(AML)中最常见的突变基因之一.LIN[27]等设计合成化合物50,在治疗急性骨髓性白血病方面较现有的FLT3抑制剂的效果更好;对血癌细胞MOLM-13异种移植小鼠有明显的抑制肿瘤生长作用,抑制率高达70%.

图26 化合物49和50的结构Fig.26 The structure of compound 49 and 50
Crizotinib为间变性淋巴瘤激酶(ALK)受体酪氨酸激酶抑制剂,在2011年获得美国食品和药物管理局认证,作为治疗ALK和ROS阳性患者的有效药物.HUANG[28]等以Crizotinib为先导化合物,通过构效关系研究合成化合物51,其在临床前期表现出明显的药代动力学特征和高效的抑制肿瘤生长的活性,活性远高于Crizotinib,其结构如图27所示:
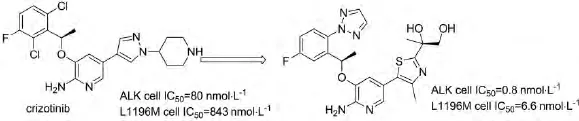
图27 化合物51的结构及活性Fig.27 The structure of compound 51 and the activity
6 结语
蛋白酪氨酸激酶在细胞的恶性生长和增殖中起着非常重要的作用,在过去几年里,出现了数个以蛋白酪氨酸激酶为靶向的抗肿瘤药物,这些药物在肿瘤治疗中具有高选择性、低毒性的治疗效果,克服了传统抗肿瘤药难以避免的选择性差、不良反应等缺点,因此设计合成高效、低毒、特异性强的小分子酪氨酸激酶抑制剂仍是抗肿瘤新药物研究开发的重要方向.
[1]RUENRAROENGSAK P,COOK J M,FLORENCE A T.Nanosystem drug targeting:facing up to complex realities[J].J Con Re,2010,141(3):265-276.
[2]茆勇军,李海泓,李剑峰,等.蛋白酪氨酸激酶信号转导途径与抗肿瘤药物 [J].药学报,2008,43(4):323-334.
[3]张志华,黄 彦,王晓明,等.小分子靶向抗癌药物-蛋白酪氨酸激酶抑制剂研究进展 [J].药学实践,2006,6(51):50-54.
[4]刘 靖,王 林,杨晓明.多靶点蛋白酪氨酸激酶抑制剂的研究进展 [J].国际医学研究杂志,2009,36(3):161-171.
[5]KLUTCHKO S R,ZHOU Haiyong,WINTER T,et al.Tyrosine kinase inhibitors.19.6-alkynamides of 4-anilinoquinazolines and 4-anilinopyrido[3,4-d]pyrimidines as irreversible inhibitors of the erbB family of tyrosine kinase receptors[J].J Med Chem,2006,49(4):1475-1485.
[6]HU E,TASKER A,WHITE R D,et al.Discovery of aryl aminoquinazoline pyridones as potent,selective,and orally efficacious inhibitors of receptor tyrosine kinase c-Kit[J].J Med Chem,2008,51(11):3065-3069.
[7]VANBROCKLIN H F,LIM J K,COFFING S L,et al.Anilinodialkoxyquinazolines:screening epidermal growth factor receptor tyrosine kinase inhibitors for potential tumor imaging probes[J].J Med Chem,2005,48(23):7445-7456.
[8]BANERJEE R,RACHID Z,MCNAMEE J,et al.Synthesis of a prodrug designed to release multiple inhibitors of the epidermal growth factor receptor tyrosine kinase and an alkylating agent:a novel tumor targeting concept[J].J Med Chem,2003,46(25):5546-5551.
[9]陈 重,李 钦,樊后兴.VEGFR-2酪氨酸激酶抑制剂3-取代吲哚-2-酮的合成及抑制活性[J].河南大学学报:自然科学版,2009,28(2):110-114.
[10]SUN Li,LIANG C,SHIRAZINA S,et al.Discovery of 5-[5-Fluoro-2-oxo-1,2-dihydroindol-(3Z)-ylidenemethyl]-2,4-dimethyl-1H-pyrrole-3-carboxylic acid(2-diethylaminoethyl)amide,a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase[J].J Med Chem,2003,46(7):1116-1119.
[11]MAHBOOBI S,UECKER A,SELLMER A,et al.Novel bis(1H-indol-2-yl)methan-ones as potent inhibitors of FLT3 and platelet-derived growth factor receptor tyrosine kinase[J].J Med Chem,2006,49(11):3101-3115.
[12]史祥飞,薛 阳,赵砚瑾,等.新型喹啉类蛋白酪氨酸激酶抑制剂的合成 [J].解放军药学学报,2011,27(3):189-192.
[13]KUBO K ,SHIMIZU T,OHYAMA S,et al.Novel potent orally active selective VEGFR-2tyrosine kinase inhibitors:synthesis,structure-activity relationships,and antitumor activities of N-phenyl-N′-{4-(4-quinolyloxy)phenyl}ureas[J].J Med Chem,2005,48(5):1359-1368.
[14]LIU F Y.Discovery of a selective irreversible BMX inhibitor for prostate cancer[J].Chem Bio,2013,8(7):1423-1428.
[15]FREY R R,CURTIN M L,ALBERT D H,et al.7-aminopyrazolo[1,5-a]pyrimidines as potent multitargeted receptor tyrosine kinase inhibitors[J].J Med Chem,2008,51(13):3777-3787.
[16]DAI Yujia,GUO Yan,FREY R R,et al.Thienopyrimidine ureas as novel and potent multi-targeted receptor tyrosine kinase inhibitors[J].J Med Chem,2005,48(19):6066-6083.
[17]HURLEY L.H,MAHADEVAN D,HAN H,et al.Protein kinase inhibitors.US20050239-794[P].2005-10-27.
[18]BRIDGES A J,DENNY W A,FRY D,et al.Tricyclic compounds capable of inhibiting tyrosine kineses of the epidermal growth factor receptor family.WO199519970[P].1995-07-27.
[19]GANGNJEE A,KURUP S,IHANT M A,et al.N4-aryl-6-substitutedphenylmethyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamines as receptor tyrosine kinase inhibitors[J].Bio Med Chem,2012(20):910-914.
[20]LANG Xuliang,SUN Qinsheng,CHEN Yuzong,et al.Novel synthetic 9-benzyloxyacridine analogue as both tyrosine kinase and topoisomerase I inhibitor[J].Chi Chem Lett,2013(24):677-680.
[21]ABDEL-MAGID A F.Inhibitors of bruton′s tyrosine kinase(Btk)may treat inflammation,immunological disorders,and cancer[J].ACS Med Chem Lett,2013,4(8):690-691.
[22]BAINDUR N ,CHADHA N,BRANDT B M,et al.2-Hydroxy-4,6-diamino-[1,3,5]triazines:a novel class of VEGF-R2(KDR)tyrosine kinase inhibitors[J].J Med Chem,2005,48(6):1717-1720.
[23]GINGRICH D R,REDDY D R,IQBAL M A,et al.A new class of potent vascular endo-thelial growth factor receptor tyrosine kinase inhibitors:structure-activity relationships for a series of 9-alkoxymethyl-12-(3-hydroxypropyl)indeno[2,1-a]pyrrol[3,4-c]-carbazole-5-ones and the identification of CEP-5214and its dimethylglycine ester prodrug clinical candidate CEP-7055[J].J Med Chem,2003,46(25):5375-5388.
[24]HO C Y,LUDOVICI D W,MAHAROOF U S M,et al.(6,7-Dimethoxy-2,4-dihydroindeno[1,2-c]pyrazol-3-yl)-phenylamines:platelet-derived growth factor receptor tyrosine kinase inhi-bitors with broad antiproliferative activity against tumor cells[J].J Med Chem,2005,48(26):8163-8173.
[25]COMEAU A B,CRITTON D A,PAGE R,et al.A focused library of protein tyro-sine phosphatase inhibitors[J].J Med Chem,2010,53(18):6768-6772.
[26]CHEKLER E L P,KISELYOV A S,OUYANG X H,et al.Discovery of dual VEGFR-2and tubulin inhibitors with in vivo efficacy[J].Med Chem Lett,2010,1(9):488-492.
[27]LIN W H,HSU J T A,HEIEH S Y,et al.Discovery of 3-phenyl-1H-5-pyrazolylamine derivatives containing a urea pharmacophore as potent and efficacious inhibitors of FMS-like tyrosine kinase-3(FLT3)[J].Bio Med Chem,2013(21):2856-2867.
[28]HUANG Qinghua,JOHNSON T W,BAILEY S,et al.Design of potent and selective inhibitors to overcome clinical anaplastic lymphoma kinase mutations resistant to crizotinib[J].J Med Chem,2014,57(4):1170-1187.
