采用系统的方法自动构建链烷烃高温燃烧反应机理
2014-09-17郭俊江华晓筱谈宁馨李象远
郭俊江 华晓筱 王 繁 谈宁馨,* 李象远
(1四川大学化学工程学院,成都610065; 2四川大学化学学院,成都610065)
1 Introduction
In recent years,great efforts have been invested to explore combustion processes of practical fuels for clean energy and engine design.However,it is difficult to investigate combustion of fuels by experiment alone.Combustion mechanisms play a critical role in elucidating the complex behavior and phe-nomena in combustion.A basic understanding of the combustion kinetics of practical fuels is also crucial in optimal design of engine.Practical fuels such as gasoline,diesel,jet fuel or kerosene are composed of thousands of different hydrocarbon compounds.A direct kinetic simulation of their chemical and physical properties is not feasible because of the complexity of practical fuels,and a viable method is to develop surrogate mixtures containing several kinds of representative pure compounds to simulate combustion behaviors.Surrogates for practical fuels are usually classified based on molecular structure,i.e.,normal alkanes,branched or iso-alkanes,cycloalkanes,olefins,aromatics,and oxygenated hydrocarbons.1-8To date,nalkane is always chosen as a representative in almost all suggested surrogate mixtures.n-Heptane is a primary reference fuel(PRF)used to define a research octane number of 0 and it is a common n-alkane representative for gasoline surrogates,9while iso-octane is used as a model compound for branched alkane components found particularly in gasoline and diesel.10,11n-Decane and n-dodecane are frequently chosen as surrogate components in different studies.12-17
Extensive experimental and kinetic modeling studies on combustion properties of alkanes have been reported.Shen and coworkers18,19investigated the ignition of n-alkane/air mixtures and iso-octane/air mixtures at elevated pressures.Beerer and McDonell20measured auto-ignition delay time of individual alkanes and mixtures of alkanes.More recently,Liu et al.21reported the ignition temperatures of non-premixed C3and C5-C12n-alkane flame.The ignition delay time of n-decane/oxidizer mixtures were studied by Zhukov,22Olchanski,23and,Kumar24et al..Laminar flame speed of C1-C12alkanes and various PRF mixtures were measured at atmospheric pressure.25-28Kelley et al.29reported laminar flame speed for iso-octane at pressures up to 1.0×106Pa and C5-C8n-alkanes up to 2.0×106Pa.
There have been substantial previous efforts aimed at kinetic modeling of alkanes.30-44Battin-Leclerc and coworkers32,33carried out a detailed modeling of the oxidation of n-octane and ndecane.Ranzi et al.16developed a lumped mechanism for investigating the combustion of n-decane,n-dodecane,and n-hexadecane,and the mechanism lumping reduces the size of each mechanism.A comprehensive detailed chemical reaction mechanism describing the oxidation of n-alkanes from n-octane to nhexadecane was developed by Westbrook group.1The mechanism for these n-alkanes is presented in a single detailed mechanism containing 2116 species and 8130 reactions.A high-temperature chemical kinetic model of n-alkane oxidation(JetSurF version 1.0,194 species,1459 reactions)was proposed by Wang group.34You et al.35proposed a high-temperature detailed kinetic model for the combustion of n-alkanes up to n-dodecane.Curran and coworkers36,37developed the detailed mechanisms for n-heptane and iso-octane oxidation,respectively.Computational fluid dynamics(CFD)plays a key role in designing the combustor.However,the use of CFD simulations requires accurately predictive and highly reduced models of the combustion of fuels.38These mechanisms are much too complicated to be used in CFD simulations.Some reduced mechanisms for n-alkane computation have been developed using various reduction techniques.31,39-47For example,a new skeletal mechanism which includes 36 species and 128 reactions was developed by Chang et al.48to describe the oxidation of n-alkanes from n-octane to n-hexadecane.This small-size mechanism can be easily integrated into the CFD simulation.
Although lots of mechanisms for the combustion of alkanes have been reported,a systematic approach which combines constructing,simplifying,validating,and analyzing the hightemperature combustion mechanisms of alkanes has not been proposed.In this paper,we develop such a systematic approach to construct reliable and reduced high-temperature combustion mechanisms of alkanes which would be helpful to CFD for engine design.Detailed mechanisms of combustion of alkanes usually contain numerous elementary reactions in addition to kinetic and thermodynamic parameters.Such comprehensive chemical kinetic mechanisms are very difficult to be composed manually and the use of an automatic program of mechanisms could facilitate this step significantly.Battin-Leclerc et al.49developed an automatic mechanism generation system EXGAS.Ranzi et al.50developed a computer code named MAMOX.Muharam and Warnatz51developed MOLEC to generate mechanisms for the combustion of large hydrocarbons.These mechanism generators are based on the reaction classes.Based on this rule,a mechanism generation program(named ReaxGen)7,17for the high and low temperature oxidation mechanism construction of alkanes and cycloalkanes has been developed in our research group.Detailed mechanisms of several alkanes,i.e.,n-heptane,iso-octane,n-decane,and n-dodecane,were constructed automatically using ReaxGen.These mechanisms were reduced using rate-of-production(ROP)analysis to obtain semi-detailed mechanisms,and skeletal mechanisms were achieved from the semi-detailed mechanisms based on path flux analysis(PFA)52.In order to verify the mechanisms,simulation results were compared with available experimental data on ignition delay time,laminar flame speed,and the concentration profile of important species.Reaction path analysis and sensitivity analysis were also employed to elucidate reaction pathways in the high-temperature oxidation of alkanes and reactions which are most important to ignition delay time,respectively.In the further study,this systematic approach will be adopted to construct oxidation mechanisms of other hydrocarbon fuels such as cycloalkanes,aromatic hydrocarbons,and alternative fuels.
2 Mechanism construction
2.1 Semi-detailed mechanism
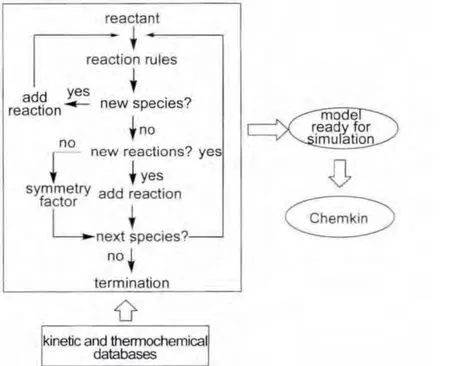
Fig.1 Primary flow chart of ReaxGen
In this work,all detailed mechanisms for the high-temperature combustion of alkanes consist of two parts,a validated core mechanism53and a sub-mechanism produced by ReaxGen.The fundamental concept in generating mechanism is the reaction class.Reaction classes are related to the species reactivity which is based solely on structural features around the reaction center.Whenever these structures are found in reactants,products can be generated.54ReaxGen mainly services for Cn(n≥5)species.In ReaxGen,the generation of a mechanism is achieved by iteratively applying a list of reaction classes to a set of molecules.First,a new set of species are obtained according to the reaction classes related to a set of initial reactant molecules.Secondly,a produced species is checked whether it is new.If this species is new,it and the reactions involved in this species are added in the reaction mechanism.If this species is not new,it is not added,and the reactions involved in this species are judged whether they are new.Then,new reactions will be added in the mechanism,while the symmetry factors of the existed reactions will be modified.In the iterative application,the reaction rules are applied to all the species until no new species are formed.Thermochemical data of species and kinetic data of reactions are automatically obtained from the databases in ReaxGen.The primary flow chart of ReaxGen is depicted in Fig.1.
The main reaction classes for the high-temperature combustion of alkanes include the following:1,36
(1)unimolecular decomposition of alkanes;
(2)H-abstraction from C atoms in alkanes by O,H,OH,O2,CH3,and HO2;
(3)mutual isomerization of the alkyl radical;
(4)decomposition of alkyl radical;
(5)oxidation of alkyl radical to form alkene;
(6)H-abstraction from the alkenes;
(7)decomposition of alkene;
(8)addition of alkenes to O,CH3,H,H2O2,and OH;
(9)decomposition of alkenyl radical.
The core mechanism(C0-C4)is very important in constructing the mechanisms of high carbon hydrocarbons.In this study,the updated USC-Mech II53,55with 111 species and 784 reactions was used as the core mechanism,which has been validated against a wide range of experimental data.53The sub-mechanisms for high carbon species Cn(n≥5)generated by ReaxGen were coupled with the C0-C4core mechanism53to construct the detailed chemical kinetic mechanisms for the high-temperature oxidation of n-heptane(271 species taking part in 1374 reactions),n-decane(615 species taking part in 2637 reactions),isooctane(432 species taking part in 1918 reactions),and n-dodecane(1008 species taking part in 4105 reactions).Thermodynamic parameters of species in sub-mechanisms were estimated based on the group additivity method,56,57while corresponding data in core mechanism were obtained from the reference.53Transport data of species were calculated through the diffusion coefficients using the approach similar to that in the reference.58In order to improve the computational efficiency,reactions and species that have negligible effect in combustion process were removed from the mechanism based on the method of ROP analysis.This method was carried out as the following:in Chemkin 2.0 output file,ROP data are the normalized contributions of each reaction to the production and consumption of each species at every time step.For obtaining the contributions of each reaction to the production and consumption of each species during the whole reaction process,ROP data were taken time to integrate over the whole reaction time through a program written by ourselves.Those reactions whose contributions to all species are less than a given threshold will be deleted.In the meantime,those species which are not involved in any reaction of the mechanism will be removed from the mechanism.This method can also be used to analyse the overall reaction path.Thus the semi-detailed mechanisms with a reasonable size can be obtained:135 species and 552 reactions for nheptane(threshold:1×10-4mol· m-3),169 species and 535 reactions for n-decane(threshold:5.5×10-4mol·m-3),202 species and 738 reactions for n-dodecane(threshold:1×10-5mol·m-3),and 115 species and 328 reactions for iso-octane(threshold:8.5×10-3mol·m-3).Our calculations show that simulation results using the semi-detailed mechanisms are almost the same as those with the detailed mechanisms.
2.2 Skeletal mechanism
Semi-detailed mechanisms were reduced to achieve skeletal mechanisms using the PFA approach.52As proposed by Lu and Law,59a two-stage reduction strategy is adequate for the reduction of large mechanisms.So in this study,a two-stage PFA reduction was implemented in order to obtain skeletal mechanisms with minimum number of species.The more details of PFA approach can be found in the reference.52rABwhich shows importance of species B to species A in PFA approach is calculated based on production and consumption of species A via B as the following:52
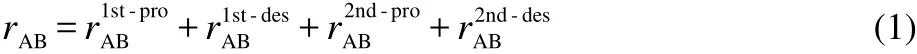
where the interaction coefficientsaredefined as:
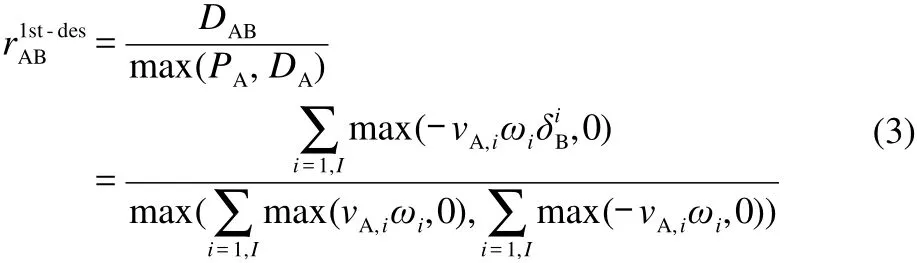
In Eqs.(2)-(3),I is the total number of elementary reactions,ωiis the reaction rate of the ith reaction,νA,iis the stoichiometric coefficient of species A in the ith reaction.If the reaction involved species B,equals to 1,otherwiseBequals to 0.PABand DABare the production and consumption rates of species A due to species B,respectively.PAand DAdenote,respectively,production and consumption fluxes of speciesA.
The interaction coefficientsoandof the second generation in Eq.(1)are the measures of flux ratios between A and B via a third reactant(Mi)for the second generation and are defined as:

Here the summation includes all possible reaction paths(fluxes)relatingAand B.To carry out mechanism reduction using PFA,a threshold value ε and a set of preselected species(e.g.,A)need to be specified.If rAB<ε,species B will be removed from the mechanism.On the other hand,species B is selected when rAB≥ ε.After deleting unimportant species and involved reactions,the skeletal mechanism can be obtained.52,60The PFA program has been implemented through an interface to Chemkin-SENKIN.61,62
In mechanism reduction,ignition delay time was chosen as the target parameter.The ignition delay time was defined as the time with the maximum value of(dxOH/dt),where xOHis the concentration(mole fraction)of the radical OH.To achieve skeletal mechanisms with wide applicability,reaction rates at points around ignition delay time under conditions of high-temperatures(1000-1600 K),high pressures(1.0×105,5.0×105,1.0×106Pa),and equivalence ratios(ϕ=0.5,1.0,2.0)from simulations using Chemkin-SENKIN61,62were used in PFA.The fuel,oxygen,and nitrogen were chosen as initial components in mechanism reduction,and numbers of species and reactions in the obtained skeletal mechanisms for these alkanes are listed in Table 1.Because adequate threshold values selected for four alkanes were different for obtaining the smallest error in reduction process,numbers of species and reactions of reduced mechanisms do not increase as the number of carbon atoms in alkanes increases.
According to our simulations,ignition delay times calculated using final skeletal mechanisms are consistent with thosebased on semi-detailed mechanisms for these alkanes.Results for n-decane are illustrated in Fig.2.One can see that simulation results of skeletal mechanisms are in good agreement with those using semi-detailed mechanisms over a wide range of parameters.
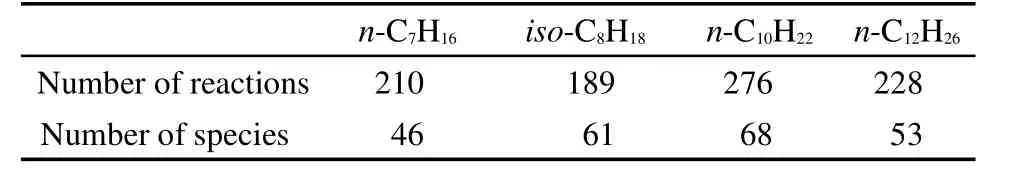
Table 1 Numbers of species and reactions in skeletal mechanisms for combustion of n-heptane,iso-octane,n-decane,and n-dodecane
3 Validation of the mechanism
Mechanism validation was fulfilled by comparing calculation results with available experimental data on ignition delay time,laminar flame speed,and the concentration profile of important species in the literature,which are highly important parameters describing global combustion properties.In this work,semi-detailed and skeletal mechanisms of n-heptane,iso-octane,n-decane,and n-dodecane were validated against ignition delay time behind reflected shock wave,laminar flame speed,and the concentration profile of important species in a jetstirred reactor(JSR).
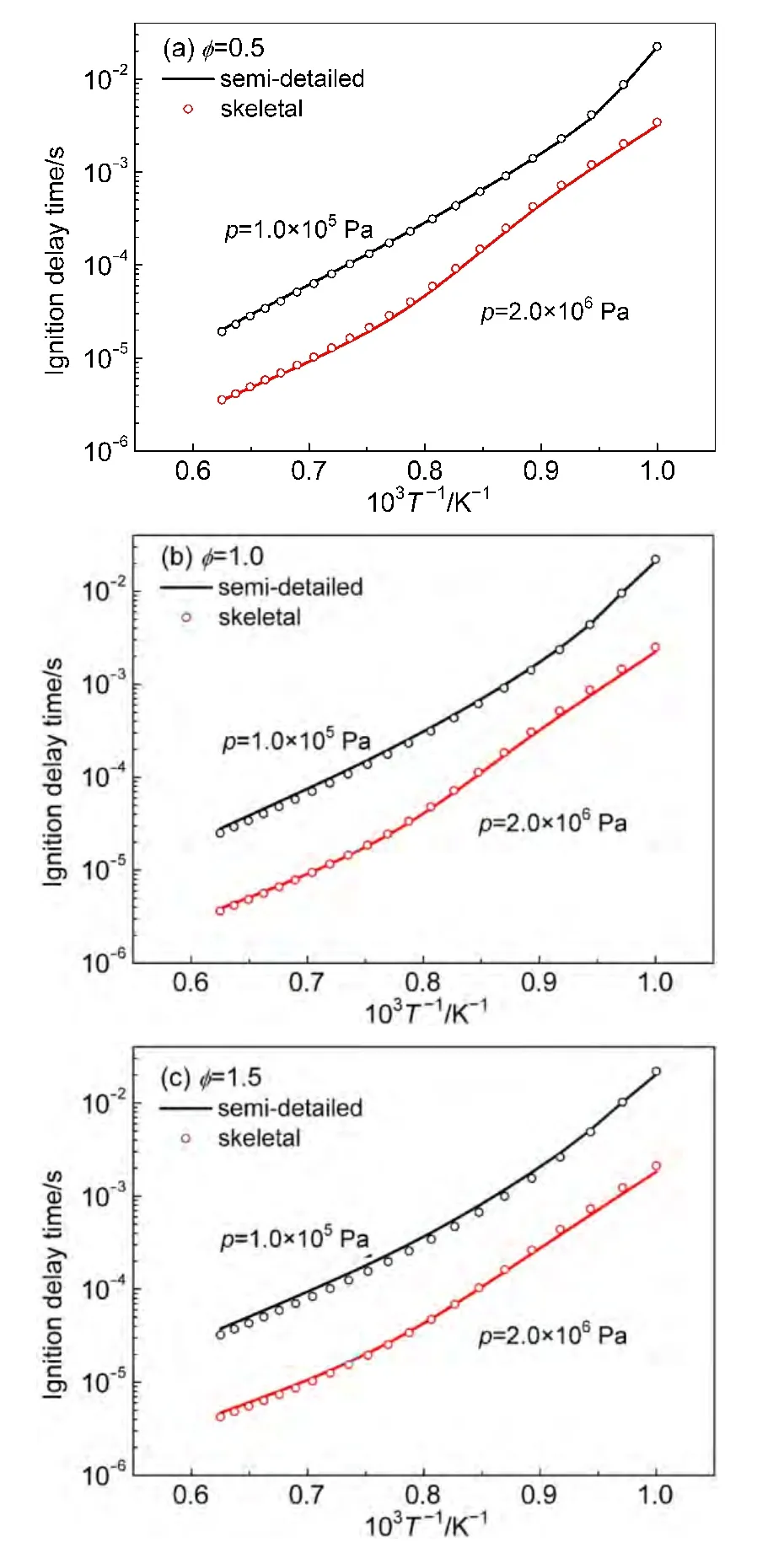
Fig.2 Igniton delay times calculated using semi-detailed and skeletal mechanisms for n-decane
3.1 Ignition delay time
The semi-detailed and skeletal mechanisms of these alkanes were employed to simulate the ignition delay times under the same experimental conditions reported by Shen and coworkers18,19using Chemkin 2.0 software package.61In addition,our results were also compared with those based on mechanisms developed by Wang34and Curran36,37et al.to demonstrate reliability of our mechanisms.Both the Curran′s n-heptane model(550 species and 2450 reactions)and iso-octane model(860 species and 3600 reactions)can be used over a wide range of temperature from 550-1700 K.Experimental ignition delay times together with the calculated ones are presented in Fig.3.Ignition delay times of n-dodecane at 1.4×106Pa versus the inverse of initial temperatures at 0.5 equivalence ratio are illustrated in Fig.3(a).Experimentally derived and numerically predicted ignition delay times for n-decane and n-heptane at 1.0 equivalence ratio and high pressures(1.2×106and 5.0×106Pa)are shown in Figs.3(b)and 3(c),respectively.The ignition delay times of iso-octane at 1.0×106Pa pressure and 1.0 equivalence ratio are given in Fig.3(d).As can be seen from Fig.3,results based on skeletal mechanisms closely resemble those using semi-detailed mechanisms,which show reliability of the skeletal mechanisms.In addition,ignition delay times for hightemperature oxidation of four kinds of alkanes obtained with our mechanisms agree reasonably well with experimental data except for those at 5.0×106Pa pressure.It can also be seen from this figure that ignition delay times from our mechanisms are generally smaller than those with other mechanisms and our results are in better agreement with experimental data.It should be noted that only high-temperature chemical reaction classes are included in our mechanisms,so they can only be applied to simulate combustions at a relatively high temperature.Moreover,compared with experimental data,ignition delay times at 5.0×106Pa pressure with our mechanisms are overestimated.This indicates that influence of pressure on kinetic and thermodynamic parameters may be important and it is also possible that alternative pathways which are not described properly in our mechanism could play a role under high pressure.
3.2 Laminar flame speed
Laminar flame speeds at 1.0×105Pa and different equivalence ratios for combustion of n-heptane,n-decane,iso-octane,and n-dodecane were calculated using the PREMIX code63coupled with the Chemkin 2.0.61Besides our models,JetSurF 1.0 developed by Wang et al.34and the model of iso-octane from Blanquart et al.64were also adopted in simulations.The Blanquart′s model including 149 species and 1651 reactions is aimed at the formation of soot precursors for fuel surrogates for premixed and diffusion.Calculated results together with available experimental data are demonstrated in Fig.4(a)for ndodecane,Fig.4(b)for n-decane,Fig.4(c)for n-heptane,and Fig.4(d)for iso-octane.Experimental values of laminar flame speeds were taken from the literature by Ji,28Kumar,26,27Kelley,29and Davis25et al.
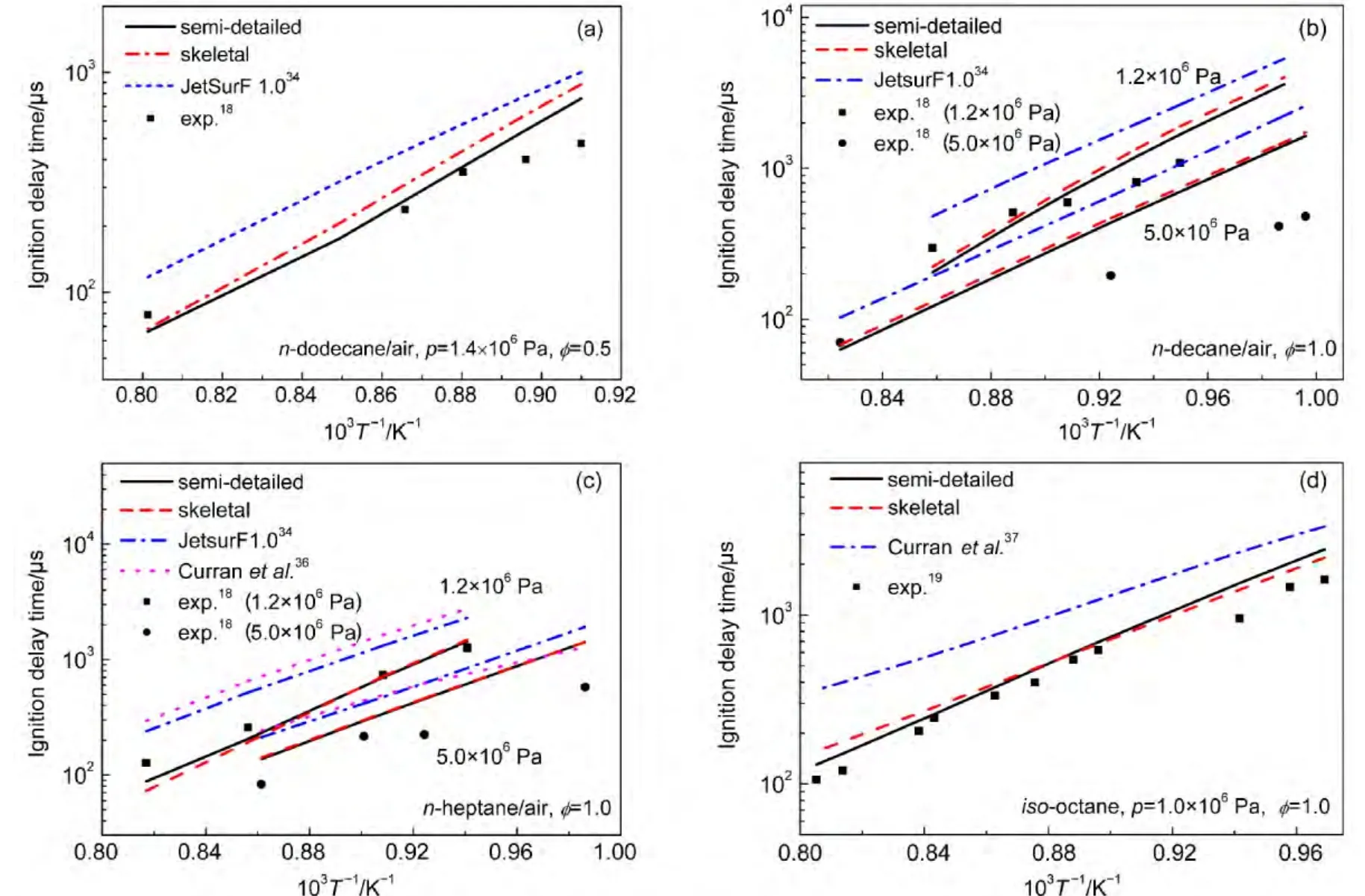
Fig.3 Ignition delay times for the high-temperature combustion of(a)n-dodecane,(b)n-decane,(c)n-heptane,(d)iso-octane
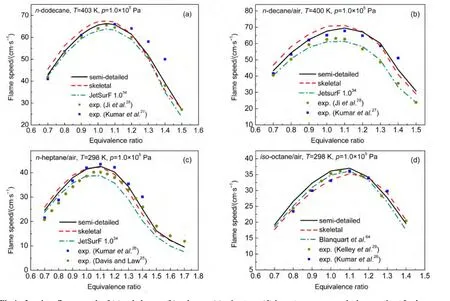
Fig.4 Laminar flame speeds of(a)n-dodecane,(b)n-decane,(c)n-heptane,(d)iso-octane versus equivalence ratio at fresh gas temperatures 403,400,and 298 K and 1.0×105Pa pressure
According to Fig.4,laminar flame speeds in present work are generally in reasonable agreement with experimental data at various equivalence ratios,and calculated results using semidetailed mechanisms agree better with experimental values than those using skeletal mechanisms.Difference in laminar flame speeds between semi-detailed mechanism and skeletal mechanism shows up and reaches 5 cm·s-1for fuel-lean mixtures,while this difference is much smaller for richest mixtures.It has been discussed previously35that flame propagation is sensitive to the rates of H2/CO/C1-C2reaction,so this difference could be a result of an oversimplification of the core mechanism.It can be seen from Fig.4 that our results are in good agreement with the experimental data of Kumar and coworkers26,27and the fuel-lean data from Ji et al.28On the other hand,laminar flame speeds using JetSurF 1.0 reaction model are smaller than our results and are more close to experimental data reported by Ji et al.28It should be noted that the data of Kumar and coworkers26,27for fuel rich mixtures are higher than those data reported by Ji et al.28,which is related to the fact that the values of Kumar and coworkers26,27were obtained by linear extrapolation while those of Ji et al.28were derived from nonlinear extrapolation.In general,results with our mechanisms agree better with experimental data derived from linear extrapolation,while results using JetSurF 1.0 are more consistent with those obtained by non-linear extrapolation.
3.3 Concentration profile of species
Dagaut and coworkers65-67have experimentally studied the oxidation of several kinds of alkanes in a JSR.The measured concentrations of many species are useful for the validation of mechanisms of alkanes.Because the consumption of reactants and production of products play a very important role during combustion,special attention is paid to the evolution of the concentrations of alkanes,oxygen,CO,CO2in this section.Simulations were performed using a zero-dimensional model under constant-pressure,isothermal conditions.
Both our semi-detailed and skeletal mechanisms of alkanes were chosen to validate the species concentration in a JSR.Simulations were implemented under the same experimental conditions reported by Dagaut and coworkers.65-67Simulation conditions were equivalence ratio ϕ=1.0,0.1%alkanes diluted in nitrogen at 1.0×106Pa,and 1.0 s residence time.Simulation results and experimental data for the mole fraction profiles of alkanes,oxygen,CO,CO2are depicted in Fig.5.The models developed by Wang(JetSurF 1.0),34Curran,36and Blanquart64et al.were also employed to simulate species concentrations under the same conditions.Simulation results and experimental data are depicted in Fig.6.
It can be seen in Figs.5-6 that all calculated concentration profiles of species using our mechanisms and published models are in qualitatively agreement with available experimental data derived from literature.65-67Simulation results from our semidetailed mechanisms are the same as those results obtained by skeletal mechanisms.That means that skeletal mechanisms maintain major reaction pathways of semi-detailed mechanisms.As the temperature is below 800 K,there are some quan-titative differences between calculated and experimental results for the species of n-dodecane,n-decane,and n-heptane oxidation using our mechanisms in Fig.5,while the same results appear in Fig.6(a,b)using JetSurF 1.0 models.The reason is that low temperature reactions have not been included in our mechanisms and JetSurF 1.0 models.The mole fraction profiles of alkanes and CO are reasonably reproduced by these models.The production of CO2is accurately reproduced by our n-do-decane model,n-dodecane model of JetSutF 1.0,Curran′s nheptane model,and Blanquart′s iso-octane model,while the consumption of oxygen is also accurately reproduced by our ndecane,n-heptane,iso-octane models,and n-decane model of Jet-SurF 1.0.However,there are still some quantitative differences between calculated results and experimental data of some species for some models,such as CO2in our n-decane model and oxygen in Curran′s n-heptane model.This result is similar to the result reported by the reference.68
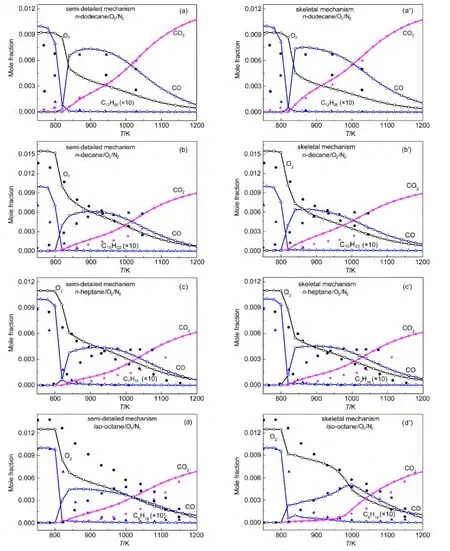
Fig.5 Chemical species concentrations simulated with our mechanisms in a JSR for 0.1%alkanesn-dodecane;(b,b′)n-decane;n-heptane;(d,d′)iso-octane)diluted in nitrogen at 1.0×106Pa pressure,ϕ=1.0,and 1.0 s residence time
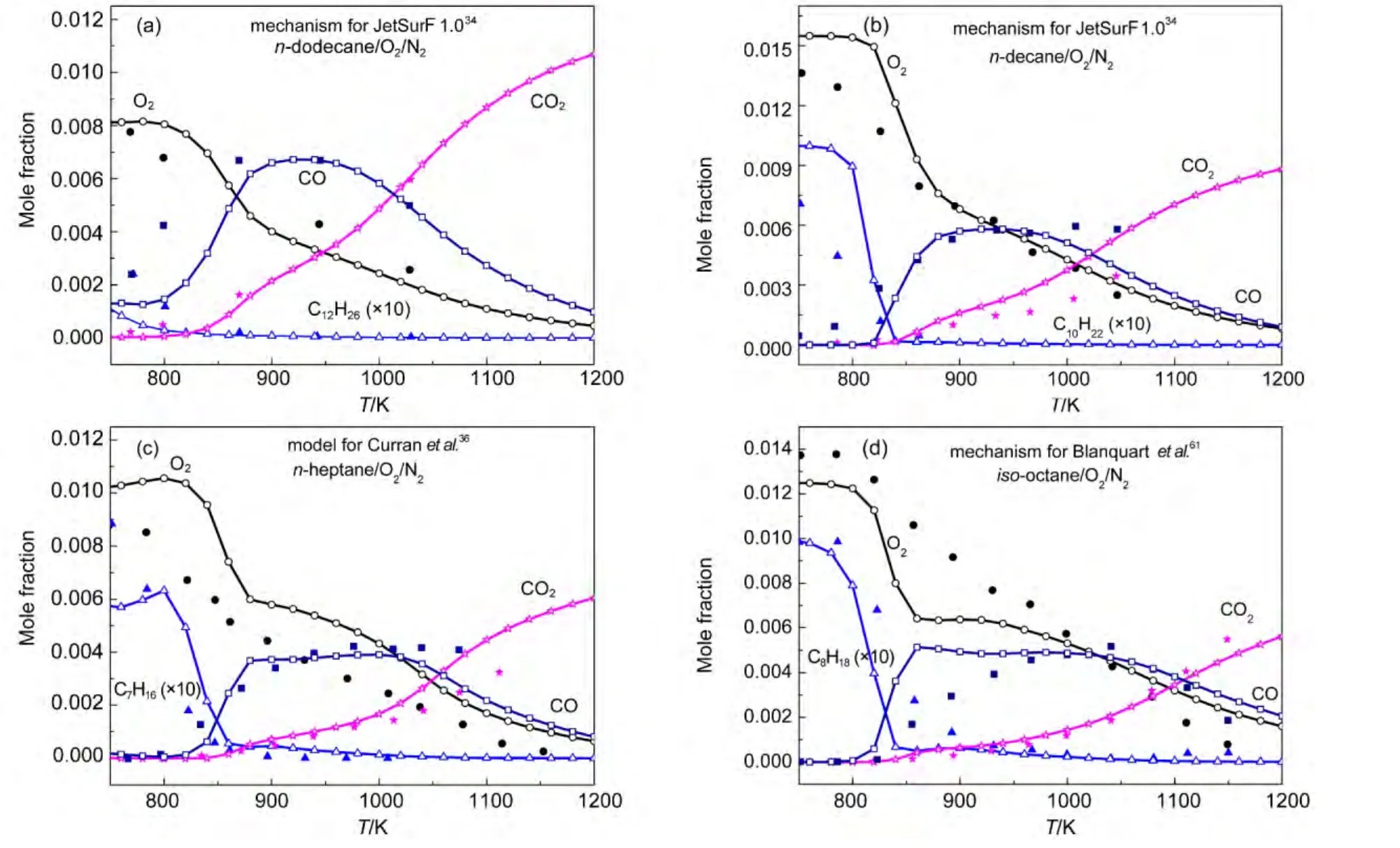
Fig.6 Chemical species concentrations simulated with published mechanisms in a JSR for 0.1%alkanes((a)n-dodecane;(b)n-decane;(c)n-heptane;(d)iso-octane))diluted in nitrogen at 1.0×106Pa pressure,ϕ=1.0,and 1.0 s residence time
In order to further verify the rationality of our mechanisms,the simulations of concentration profiles of important species in a JSR under other conditions were implemented.Simulation results and experimental data for the mole fraction profiles of alkanes,oxygen,CO,CO2are depicted in Figs.7-9.
Under the conditions of equivalence ratio ϕ=0.5,0.1%alkanes diluted in nitrogen at 1.0×106Pa pressure,and 1.0 s residence time,the simulations of concentration profiles of important species in a JSR for all alkanes except n-decane,which is short of available experimental data under the same conditions,are illustrated in Fig.7.It can be seen in Fig.7 that all calculated concentration profiles of species using our mechanisms are still in qualitatively agreement with available experimental data derived from literature.65,67Calculated results using these models show great agreement with experimental data for the formation of CO and CO2especially for n-heptane and iso-octane.The conversion of iso-octane is also excellently reproduced by the semi-detailed and skeletal mechanisms.However,there are still some quantitative differences between calculated results and experimental data of some species in the models,such as oxygen in our n-heptane and iso-octane models.The simulations for the n-decane oxidation in a JSR under the conditions,equivalence ratios ϕ=1.0 and 1.5,0.1%n-decane diluted in nitrogen at 1.0×106Pa pressure and 0.5 s residence time,were carried out in our study.Simulation results and experimental data are depicted in Figs.8-9.As can be seen from Figs.8-9,the concentrations of decane and CO2are reproduced by our semi-detailed and skeletal mechanisms.Comparing with experimental data,there are still some quantitative differences of the results for the concentrations of oxygen and COsimulatedby our n-decane model.That means the further investigation should be carried out in order to accurately reproduce all kinds of species involved in alkane oxidation.
4 Analysis of the mechanism and discussion
4.1 Reaction pathway analysis
To better understand combustion process and further optimize the combustion mechanisms of hydrocarbon fuels,reaction pathway analysis were carried out.In reaction pathway analysis,contribution of each reaction to the production and consumption of every species were first calculated using the method of ROP analysis at each time step in the whole reaction progress.Overall production and consumption reaction pathways of main species of the mechanism can be obtained by integration over the whole reaction time.The reaction pathway analysis for combustion of four alkanes based on the skeletal mechanisms was performed using the closed homogeneous batch reactor model in Chemkin 2.0 at 0.5 equivalence ratio,1×105Pa pressure,and 1150 K temperature.Main reaction routes in combustion of these alkanes are presented in Figs.10-13.
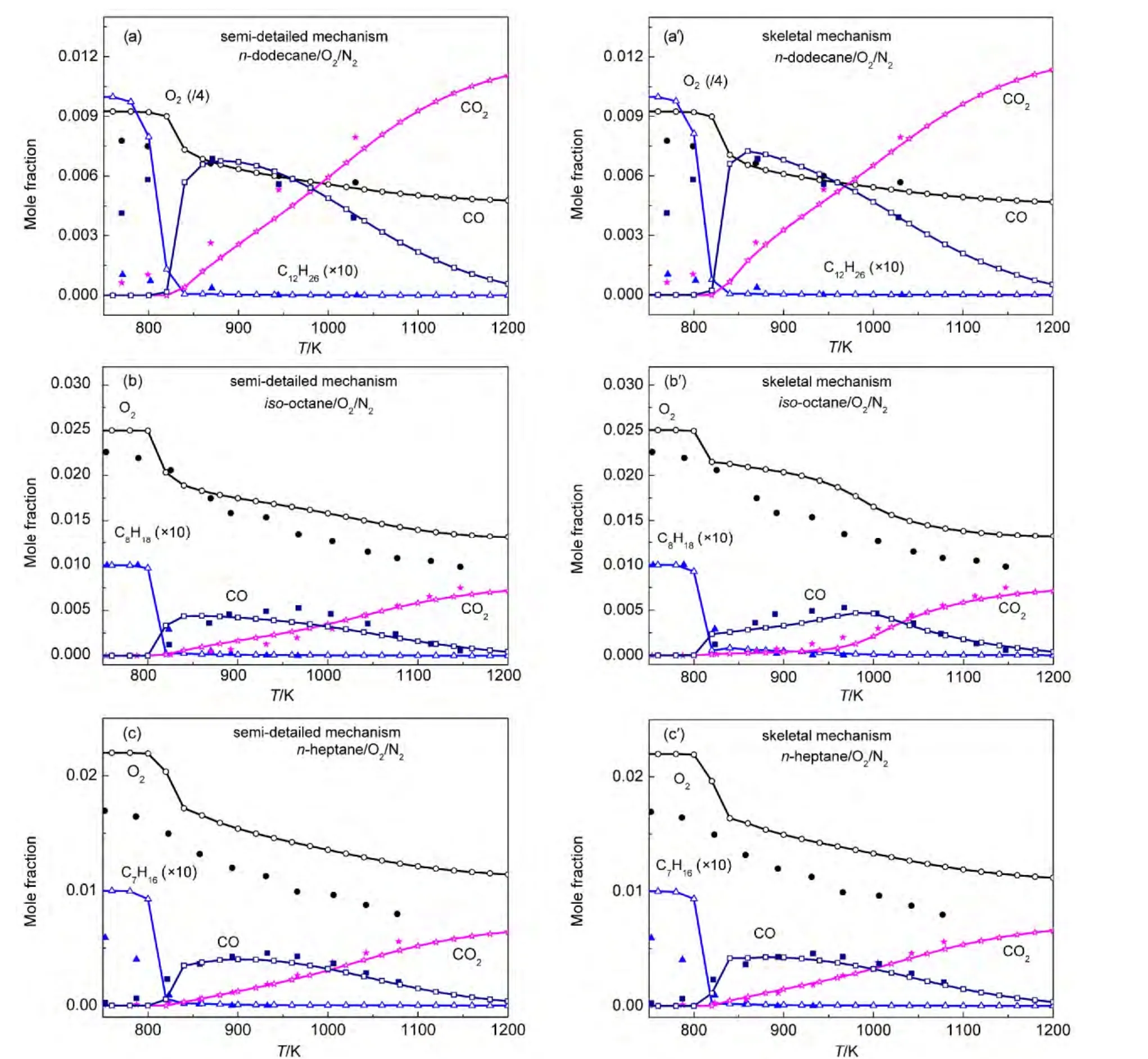
Fig.7 Chemical species concentrations in a JSR for 0.1%alkanesn-dodecane;(b,b′)iso-octane;n-heptane)diluted in nitrogen at 1.0×106Pa pressure,ϕ=0.5,and 1.0 s residence time
It can be seen from these figures that the fuels,i.e.,n-heptane,iso-octane,n-decane,and n-dodecane,are mainly consumed through H-abstraction reactions with H,OH,and O radicals(over 85%)to generate different alkyl radicals.Mutual isomerization will take place between isomers of these alkyl radicals.In addition,these alkyl radicals will undergo β-scission reactions to form olefins and smaller alkyl radicals.These smaller alkyl radicals either isomerize to form other radicals or are further consumed through β-scission reactions to produce even smaller molecules and radicals.Taken n-dodecane as an example,six different types of dodecyl radicals are generated through H-abstraction reactions.They can convert from one type to another type of dodecyl radical through isomerization or go through β-scission reactions to produce lowmolecular weight olefins and small alkyl radicals.These small alkyls can further decompose to olefin and smaller alkyls through βscission reactions.Based on comprehensive analysis of the reaction pathway in combustion of n-dodecane,we found that the consumption of 2-dodecyl radicals was the main reaction pathway.Once produced,2-dodecyl radicals decompose to propylene(C3H6)molecule and 1-nonyl(1-C9H19)radicals.1-Nonyl radicals mainly undergo isomerization to form 5-nonyl radicals and small hydrocarbon molecules(such as methane,ethylene,methyl,ethyl,1,3-butadiene)can be produced resulting from βscission reactions of 5-nonyl radicals.
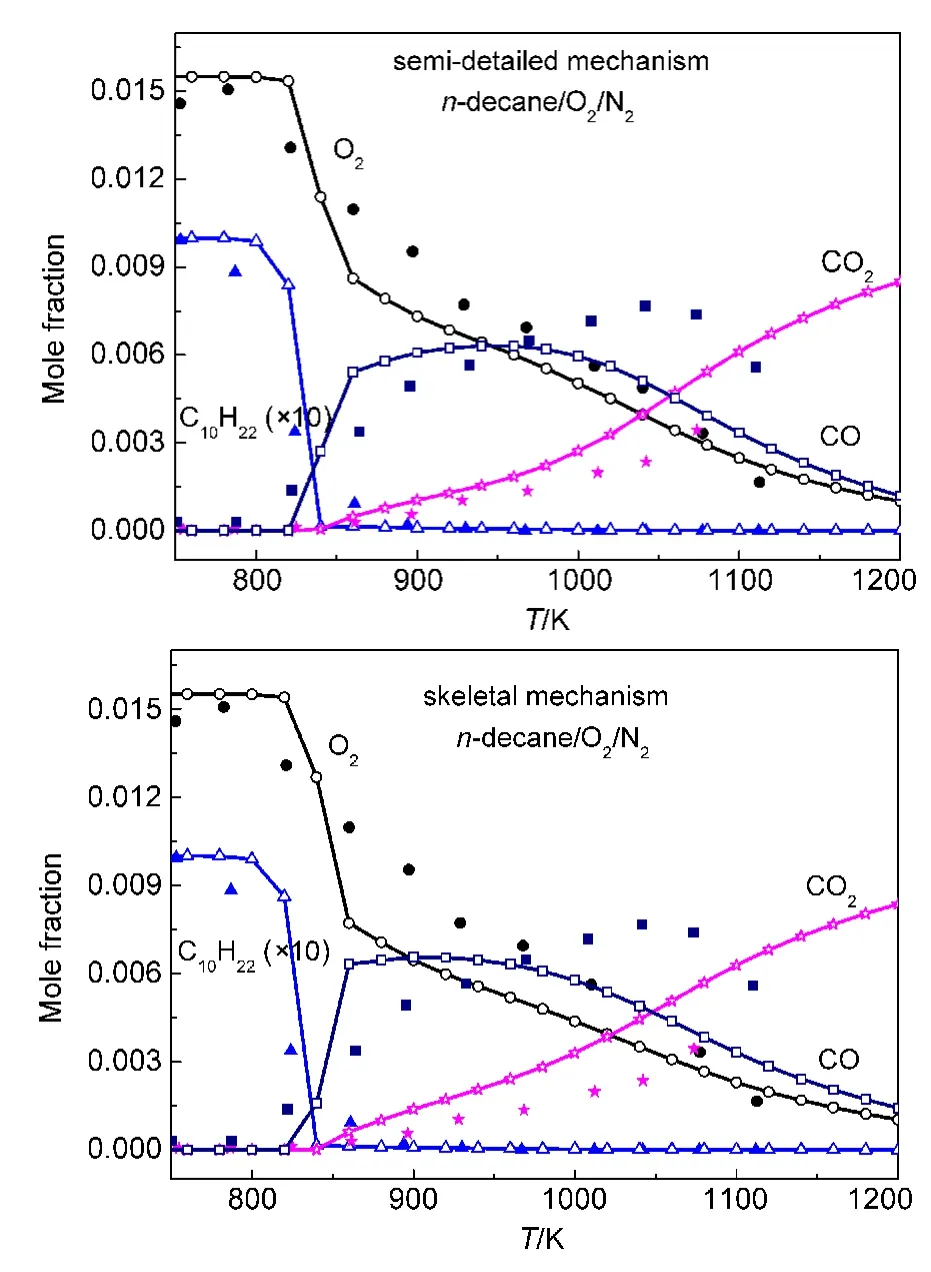
Fig.8 Chemical species concentrations in a JSR for 0.1%ndecane diluted in nitrogen at 1.0×106Pa pressure,ϕ=1.0,and 0.5 s residence time
4.2 Sensitivity analysis
In order to illustrate key reactions affecting ignition during high-temperature combustion of alkanes,sensitivity analysis for ignition delay times of n-dodecane,n-decane,n-heptane,and iso-octane were performed at ϕ=0.5,1150 K initial temperature,and 1.0×105Pa pressure.The method proposed in reference69was adopted to calculate sensitivity of the ignition delay time concerning reaction i as the following:

where τignis ignition delay ti,me calculated by the original combustion mechanism,τign(2ki)is ignition delay time simulated using this mechanism in which the rate constant of reaction i is doubled through multiplying the pre-exponential factor of reaction i by 2.A positive sensitivity implies that the related reaction has an inhibiting effect on ignition;on the contrary,a negative sensitivity indicates a promoting effect.
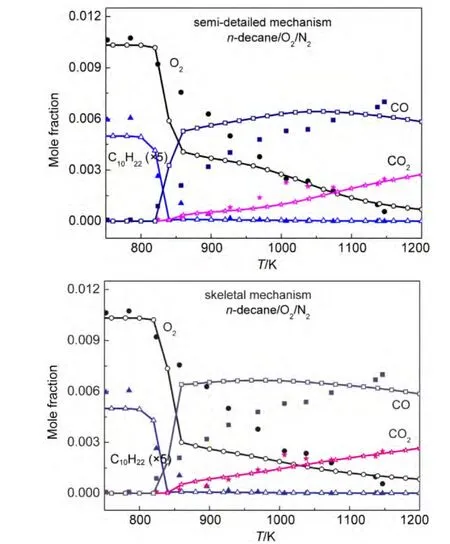
Fig.9 Chemical species concentrations in a JSR for 0.1%ndecane diluted in nitrogen at 1.0×106Pa pressure,ϕ=1.5,and 0.5 s residence time
Fig.14 demonstrates reactions that have greater effect on ignition delay times according to Eq.(6)as well as corresponding sensitivity values in our semi-detailed and skeletal mechanisms.One can see from this figure that results derived from skeletal mechanisms agree approximately with those using semi-detailed mechanisms.Furthermore,reactions in core mechanism are very important for ignition delay time,while reactions in a set of generated Cn(n≥5)sub-mechanism are less important except for iso-octane,although they are critical in initial steps in combustion.Two reactions with higher negative sensitivity values are always H+O2=OH+O and CH3+HO2=CH3O+OH during combustion of these alkanes.These two reactions thus play an important role in high-temperature chemical process.On the other hand,the reaction C2H3+O2=CHO+HCHO exhibits the largest inhibiting effect among all the reactions for straightchain alkanes,while the radical-radical combination reaction 2CH3(+M)=C2H6(+M)has the largest positive sensitivity value for the branched alkane iso-octane.Moreover,reactions involving HO2/H/OH/CH3/C2H3radicals have large influence on ignition delay time prediction at high temperature for straightchain alkanes.For iso-octane,reactions(H+O2=OH+O and CH3+HO2=CH3O+OH)as well as the fuel-consuming reactions involved the breaking of C―C bond(i-C8H18→CH3+i-C7H15-2,1-i-C8H17→i-C4H9+i-C4H8,i-C8H18→t-C4H9+i-C4H9and i-C8H17-4→CH3+i-C7H14-4)also play an important role in ignition of iso-octane at high temperature.
It is noted that the sensitivity absolute values of several reac-
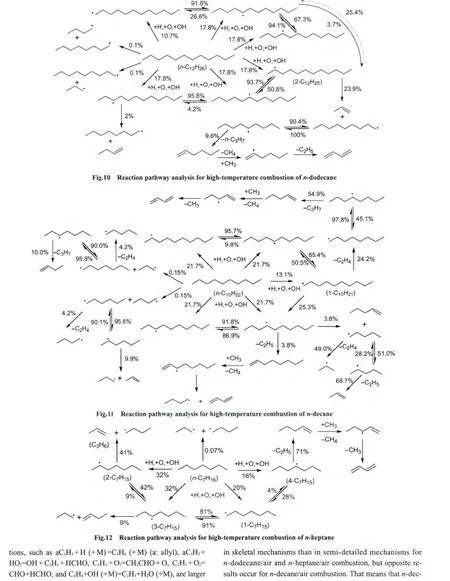
those above mentioned reactions are larger in the skeletal mechanisms than in the semi-detailed mechanisms for n-dodecane/air and n-heptane/air combustion.On the contrary,one can draw the conclusion that the consumption of 1-decatyl radicals is the main reaction pathway for n-decane from Fig.11.It is ethene not propylene that is produced through the decomposition of 1-decatyl.So the sensitivity absolute values of reactions involved in the consumption of propylene are smaller in the skeletal mechanism than in the semi-detailed mechanism for n-decane.But it is noted that the sensitivity absolute value of reaction involved in the consumption of ethene C2H4+OH=C2H3+H2O is also smaller in the skeletal mechanism than in the semi-detailed mechanism for n-decane.Because the ignition delay time used in this work is defined as the time when the largest changing rate(dxOH/dt)of OH radical concentration occurs,more ethenes produced from 1-decatyl decomposition react with more OH radicals,and then this will result in a consequence that the sensitivity value of the reaction C2H4+OH=C2H3+H2O shifts towards inhibited ignition in the skeletal mechanism.
5 Conclusions
Detailed mechanisms for high-temperature combustion of the following alkanes:n-heptane,n-decane,n-dodecane,and iso-octane were generated using the automatic mechanism generation program(ReaxGen).Because of the complexity of detailed mechanisms,these mechanisms were reduced first by employing the method of rate-of-production analysis to generate semi-detailed mechanisms.Highly reduced skeletal mechanisms were subsequently obtained using path flux analysis based on the semi-detailed mechanisms.Our results demonstrate that the skeletal mechanisms can accurately and comprehensively reproduce results of the detailed mechanisms.These mechanisms were validated against experimental data over a wide range of conditions to investigate their reliability.Simulation results show that these mechanisms are able to provide a reasonable prediction on ignition delay time,laminar flame speed,and the concentration profile of species.This indicates that the detailed and skeletal mechanisms are reliable in describing combustion behaviors of these alkanes under various conditions.Main pathways in combustion process of the alkanes at high temperature were illustrated based on the reaction path analysis.Furthermore,sensitivity analysis was also carried out,and our results indicate that reactions involving small molecules and radicals in core mechanism have great influence on ignition delay time.The reaction of H+O2=OH+O is found to be the most important reaction to promote the ignition during high-temperature combustion.
Our results show that combustion mechanisms for these alkanes,which were produced based on reaction classes and a given core mechanism,are reliable in describing high-temperature combustion process.Skeletal mechanisms with smaller numbers of species and reactions could also be helpful in understanding reaction processes and in computational fluid dynamics for engine design.Moreover,the method,which combines constructing,simplifying,validating,and analyzing the high-temperature combustion mechanisms of alkanes,could also be used to generate the mechanism of other hydrocarbons for high-temperature combustion.Mechanisms for low-temperature combustion are also of great importance and it will be desirable to be generated automatically.Work in this direction is in progress.
Supporting Information: The input files with Chemkin format about mechanisms of n-heptane,iso-octane,n-decane,and n-dodecane are available free of charge via the internet at http://www.whxb.pku.edu.cn and http://www.ccg.scu.edu.cn.
(1) Westbrook,C.K.;Pitz,W.J.;Herbinet,O.;Curran,H.J.;Silke,E.J.Combust.Flame 2009,156,181.doi:10.1016/j.combustflame.2008.07.014
(2) Oehlschlaeger,M.A.;Steinberg,J.;Westbrook,C.K.;Pitz,W.J.Combust.Flame 2009,156,2165.doi:10.1016/j.combustflame.2009.05.007
(3)Mehl,M.;Vanhove,G.;Pitz,W.J.;Ranzi,E.Combust.Flame 2008,155,756.doi:10.1016/j.combustflame.2008.07.004
(4) Mehl,M.;Pitz,W.J.;Westbrook,C.K.;Yasunaga,K.;Conroy,C.;Curran,H.J.Proc.Combust.Inst.2011,33,201.doi:10.1016/j.proci.2010.05.040
(5) Mehl,M.;Pitz,W.J.;Westbrook,C.K.;Curran,H.J.Proc.Combust.Inst.2011,33,193.doi:10.1016/j.proci.2010.05.027
(6) Oehlschlaeger,M.A.;Shen,H.P.S.;Frassoldati,A.;Pierucci,S.;Ranzi,E.Energy Fuels 2009,23,1464.doi:10.1021/ef800892y
(7)Tan,N.X.;Wang,J.B.;Hua,X.X.;Li,Z.R.;Li,X.Y.Chem.J.Chin.Univ.2011,32,1832.[谈宁馨,王静波,华晓筱,李泽荣,李象远.高等学校化学学报,2011,32,1832.]
(8) Yao,T.;Zhong,B.J.Acta.Phys.-Chim.Sin.2013,29,237.[姚 通,钟北京.物理化学学报,2013,29,237.]doi:10.3866/PKU.WHXB201211271
(9) Andrae,J.C.G.;Björnbom,P.;Cracknell,R.F.;Kalghatgi,G.T.Combust.Flame 2007,149,2.doi:10.1016/j.combustflame.2006.12.014
(10) Zheng,D.;Zhong,B.J.Acta.Phys.-Chim.Sin.2012,28,2029.[郑 东,钟北京.物理化学学报,2012,28,2029.]doi:10.3866/PKU.WHXB201207042
(11) Pang,B.;Xie,M.Z.;Jia,M.;Liu,Y.D.Acta.Phys.-Chim.Sin.2013,29,2523.[庞 斌,谢茂昭,贾 明,刘耀东.物理化学学报,2013,29,2523.]doi:10.3866/PKU.WHXB201310161
(12) Jahangirian,S.;McEnally,C.S.;Gomez,A.Combust.Flame 2009,156,1799.doi:10.1016/j.combustflame.2009.03.003
(13) Honnet,S.;Seshadri,K.;Niemann,U.;Peters,N.Proc.Combust.Inst.2009,32,485.doi:10.1016/j.proci.2008.06.218
(14) Natelson,R.H.;Kurman,M.S.;Cernansky,N.P.;Miller,D.L.Fuel 2008,87,2339.doi:10.1016/j.fuel.2007.11.009
(15) Dagaut,P.;Bakali,A.E.;Ristori,A.Fuel 2006,85,944.doi:10.1016/j.fuel.2005.10.008
(16) Ranzi,E.;Frassoldati,A.;Granata,S.;Faravelli,T.Ind.Eng.Chem.Res.2005,44,5170.
(17)Hua,X.X.;Wang,J.B.;Wang,Q.D.;Tan,N.X.;Li,X.Y.Acta Phys.-Chim.Sin.2011,27,2755.[华晓筱,王静波,王全德,谈宁馨,李象远.物理化学学报,2011,27,2755.]doi:10.3866/PKU.WHXB20112755
(18) Shen,H.P.S.;Steinberg,J.;Vanderover,J.;Oehlschlaeger,M.A.Energy Fuels 2009,23,2482.doi:10.1021/ef8011036
(19) Shen,H.P.S.;Vanderover,J.;Oehlschlaeger,M.A.Combust.Flame 2008,155,739.doi:10.1016/j.combustflame.2008.06.001
(20) Beerer,D.J.;McDonell,V.G.Proc.Combust.Inst.2011,33,301.doi:10.1016/j.proci.2010.05.015
(21) Liu,N.;Ji,C.;Egolfopoulos,F.N.Combust.Flame 2012,159,465.doi:10.1016/j.combustflame.2011.07.012
(22) Zhukov,V.P.;Sechenov,V.A.;Starikovskii,A.Y.Combust.Flame 2008,153,130.doi:10.1016/j.combustflame.2007.09.006
(23) Olchanski,E.;Burcat,A.Int.J.Chem.Kinet.2006,38,703.doi:10.1002/kin.20204
(24) Kumar,K.;Mittal,G.;Sung,C.J.Combust.Flame 2009,156,1278.doi:10.1016/j.combustflame.2009.01.009
(25) Davis,S.G.;Law,C.K.Combust.Sci.Technol.1998,140,427.doi:10.1080/00102209808915781
(26) Kumar,K.;Freeh,J.E.;Sung,C.J.;Huang,Y.J.Propul.Power 2007,23,428.doi:10.2514/1.24391
(27) Kumar,K.;Sung,C.J.Combust.Flame 2007,151,209.doi:10.1016/j.combustflame.2007.05.002
(28) Ji,C.;Dames,E.;Wang,Y.L.;Wang,H.;Egolfopoulos,F.N.Combust.Flame 2010,157,277.doi:10.1016/j.combustflame.2009.06.011
(29) Kelley,A.P.;Liu,W.;Xin,Y.X.;Smallbone,A.J.;Law,C.K.Proc.Combust.Inst.2011,33,501.doi:10.1016/j.proci.2010.05.058
(30) Jahangirian,S.;Dooley,S.;Haas,F.M.;Dryer,F.L.Combust.Flame 2012,159,30.doi:10.1016/j.combustflame.2011.07.002
(31) Sheen,D.A.;Wang,H.Combust.Flame 2011,158,645.doi:10.1016/j.combustflame.2010.12.016
(32) Glaude,P.A.;Warth,V.;Fournet,R.;Battin-Leclerc,F.;Scacchi,G.;Côme,G.M.Int.J.Chem.Kinet.1998,30,949.doi:10.1002/(SICI)1097-4601(1998)30:12<949::AID-KIN10>3.0.CO;2-G
(33) Battin-Leclerc,F.Prog.Energy Combust.Sci.2008,34,440.doi:10.1016/j.pecs.2007.10.002
(34) Sirjean,B.;Dames,E.;Sheen,D.A.;You,X.Q.;Sung,C.;Holley,A.T.;Egolfopoulos,F.N.;Wang,H.;Vasu,S.S.;Davidson,D.F.;Hanson,R.K.;Pitsch,H.;Bowman,C.T.;Kelley,A.;Law,C.K.;Tsang,W.;Cernansky,N.P.;Miller,D.L.;Violi,A.;Lindstedt,R.P.A High-Temperature Chemical Kinetic Model of n-Alkane Oxidation,JetSurF Version 1.0.http://melchior.usc.edu/JetSurF/Version1_0/index.html(accessed September 15,2009).
(35)You,X.;Egolfopoulos,F.N.;Wang,H.Proc.Combust.Inst.2009,32,403.doi:10.1016/j.proci.2008.06.041
(36) Curran,H.J.;Gaffuri,P.;Pitz,W.J.;Westbrook,C.K Combust.Flame 1998,114,149.doi:10.1016/S0010-2180(97)00282-4
(37) Curran,H.J.;Gaffuri,P.;Pitz,W.J.;Westbrook,C.K.Combust.Flame 2002,129,253.doi:10.1016/S0010-2180(01)00373-X
(38)Wang,Q.D.;Wang,J.B.;Li,J.Q.;Tan,N.X.;Li,X.Y.Combust.Flame 2011,158,217.doi:10.1016/j.combustflame.2010.08.010
(39) Wen,F.;Zhong,B.J.Acta Phys.-Chim.Sin.2012,28,1306.[文 斐,钟北京.物理化学学报,2012,28,1306.]doi:10.3866/PKU.WHXB201204012
(40)Zeuch,T.;Moréac,G.;Ahmed,S.S.;Mauss,F.Combust.Flame 2008,155,651.doi:10.1016/j.combustflame.2008.05.007
(41) Lu,T.;Law,C.K.Combust.Flame 2008,154,153.doi:10.1016/j.combustflame.2007.11.013
(42) Sarathy,S.M.;Westbrook,C.K.;Mehl,M.;Pitz,W.J.;Togbe,C.;Dagaut,P.;Wang,H.;Oehlschlaeger,M.A.;Niemann,U.;Seshadri,K.;Veloo,P.S.;Ji,C.;Egolfopoulos,F.N.;Lu,T.Combust.Flame 2011,158,2338.doi:10.1016/j.combustflame.2011.05.007
(43)Fang,Y.M.;Wang,Q.D.;Wang,F.;Li,X.Y.Acta Phys.-Chim.Sin.2012,28,2536.[方亚梅,王全德,王 繁,李象远.物理化学学报,2012,28,2536.]doi:10.3866/PKU.WHXB201208201
(44) Bikas,G.;Peters,N.Combust.Flame 2001,126,1456.doi:10.1016/S0010-2180(01)00254-1
(45) Zeppieri,S.P.;Klotz,S.D.;Dryer,F.L.Proc.Combust.Inst.2000,28,1587.doi:10.1016/S0082-0784(00)80556-1
(46) Zhong,B.J.;Yao,T.;Wen,F.Acta Phys.-Chim.Sin.2014,30,210.[钟北京,姚 通,文 斐.物理化学学报,2014,30,210.]doi:10.3866/PKU.WHXB201312103
(47) Jiang,Y.;Qiu,R.Acta Phys.-Chim.Sin.2009,25,1019.[蒋 勇,邱 榕.物理化学学报,2009,25,1019.]doi:10.3866/PKU.WHXB20090426
(48) Chang,Y.;Jia,M.;Liu,Y.;Li,Y.;Xie,M.;Yin,H.Energy Fuels 2013,27,3467.doi:10.1021/ef400460d
(49) Warth,V.;Battin-Leclerc,F.;Fournet,R.;Glaude,P.A.;Côme,G.M.;Scacchi,G.Comput.Chem.2000,24,541.doi:10.1016/S0097-8485(99)00092-3
(50) Ranzi,E.;Faravelli,T.;Gaffuri,P.;Garavaglia,E.;Goldaniga,A.Ind.Eng.Chem.Res.1997,36,3336.doi:10.1021/ie960603c
(51) Muharam,Y.;Warnatz,J.Phys.Chem.Chem.Phys.2007,9,4218.doi:10.1039/b703415f
(52) Sun,W.;Chen,Z.;Gou,X.;Ju,Y.Combust.Flame 2010,157,1298.doi:10.1016/j.combustflame.2010.03.006
(53) Wang,H.;You,X.Q.;Joshi,A.V.;Davis,S.G.;Laskin,A.;Egolfopoulos,F.N.;Law,C.K.USC Mech Version II.High-Temperature Combustion Reaction Model of H2/CO/C1-C4 Compounds.http://ignis.usc.edu/USC_Mech_II.htm(accessed May,2007).
(54) Moreác,G.;Blurock,E.S.;Mauss,F.Combust.Sci.Technol.2006,178,2025.doi:10.1080/00102200600793262
(55)Davis,S.G.;Law,C.K.;Wang,H.Combust.Flame 1999,119,375.doi:10.1016/S0010-2180(99)00070-X
(56) Benson,S.W.Thermochemical Kinetics,2nd ed.;John Wiley and Sons:New York,1976;pp 19-72.
(57) Lay,T.H.;Bozzelli,J.W.;Dean,A.M.;Ritter,E.R.J.Phys.Chem.1995,99,14514.doi:10.1021/j100039a045
(58)Wang,H.;Frenklach,M.Combust.Flame 1994,96,163.doi:10.1016/0010-2180(94)90167-8
(59) Lu,T.;Law,C.K.Combust.Flame 2006,144,24.doi:10.1016/j.combustflame.2005.02.015
(60)Wang,Q.D.;Fang,Y.M.;Wang,F.;Li,X.Y.Combust.Flame 2012,159,91.doi:10.1016/j.combustflame.2011.05.019
(61) Kee,R.J.;Rupley,F.M.;Miller,J.A.Chemkin-II:a Fortran Chemical Kinetics Package for the Analysis of Gas-Phase Chemical Kinetics.Report SAND89-8009,Sandia,1989.
(62) Lutz,A.E.;Kee,R.J.;Miller,J.A.SENKIN:a Fortran Program for Predicting Homogeneous Gas Phase Chemical Kinetics with Sensitivity Analysis.Report SAND87-8248,Sandia,1987.
(63)Kee,R.J.;Grcar,J.F.;Smooke,M.D.;Miller,J.A.PREMIX:a Fortran Program for Modeling Steady Laminar One-Dimensional Premixed Flames.Report SAND85-8240,Sandia,1985.
(64) Blanquart,G.;Pepiot-Desjardins,P.;Pitsch,H.Combust.Flame 2009,156,588.doi:10.1016/j.combustflame.2008.12.007
(65) Dagaut,P.;Reuillon,M.;Cathonnet,M.Combust.Sci.Technol.1994,95,233.
(66) Dagaut,P.;Reuillon,M.;Cathonnet,M.Combust.Sci.Technol.1994,103,349.doi:10.1080/00102209408907703
(67)Mzé-Ahmed,A.;Hadj-Ali,K.;Dagaut,P.;Dayma,G.Energy Fuels 2012,26,4253.doi:10.1021/ef300588j
(68) Chang,Y.;Jia,M.;Liu,Y.;Li,Y.;Xie,M.Combust.Flame 2013,160,1315.doi:10.1016/j.combustflame.2013.02.017
(69)Kumar,K.;Mittal,G.;Sung,C.J.;Law,C.K.Combust.Flame 2008,153,343.doi:10.1016/j.combustflame.2007.11.012
